Engineering the Future: How Advanced Protein Design Unlocks New Photoenzyme Functions with Visible Light
This article provides researchers, scientists, and drug development professionals with a comprehensive overview of the transformative advances in protein engineering for creating new photoenzyme functions.
Engineering the Future: How Advanced Protein Design Unlocks New Photoenzyme Functions with Visible Light
Abstract
This article provides researchers, scientists, and drug development professionals with a comprehensive overview of the transformative advances in protein engineering for creating new photoenzyme functions. It explores the foundational shift from UV-dependent systems to visible-light-powered biocatalysts using genetically encoded sensitizers like thioxanthone[citation:1]. The scope covers core methodological breakthroughs in directed evolution and computational design for activities such as enantioselective cycloadditions and radical C–C couplings[citation:1][citation:5]. It details strategies for troubleshooting challenges like oxygen sensitivity and spectral tuning, including the engineering of photoenzymes that operate with red light[citation:2]. Finally, the article validates these engineered systems by comparing their efficiency, selectivity, and industrial potential against traditional small-molecule photocatalysts and earlier enzymatic platforms, highlighting their ability to perform demanding syntheses of drug-relevant scaffolds in air[citation:1][citation:3].
From UV to Visible Light: Laying the Groundwork for Next-Generation Photoenzymes
This whitepaper is framed within a broader thesis on protein engineering for new photoenzyme functions. The central premise is that the strategic fusion of natural enzymatic catalysis with photochemical principles represents a frontier for creating novel biocatalysts. These engineered photoenzymes offer unprecedented spatiotemporal control over chemical reactions, with profound implications for synthetic biology, chemical manufacturing, and targeted drug development. This document serves as a technical guide to the core concepts, current data, and experimental methodologies defining this rapidly advancing field.
Fundamental Principles and Natural Photoenzymes
Photoenzymes are defined as enzymes that utilize light as a source of energy or as an essential cofactor to catalyze a chemical transformation. Natural systems provide the blueprint, with a limited but impactful set of known native photoenzymes.
Table 1: Characterized Natural Photoenzymes and Core Functions
| Enzyme | EC Number | Reaction Catalyzed | Cofactor/Chromophore | Primary Function in Nature |
|---|---|---|---|---|
| DNA Photolyase | EC 4.1.99.3 | Light-dependent repair of cyclobutane pyrimidine dimers (CPDs) | FAD, MTHF/8-HDF | DNA repair |
| (6-4) Photolyase | EC 4.1.99.3 | Repair of pyrimidine-(6-4)-pyrimidone photoproducts | FAD, MTHF/8-HDF | DNA repair |
| Fatty Acid Photodecarboxylase (FAP) | EC 4.1.1.- | Light-driven decarboxylation of fatty acids to alkanes | FAD | Aliphatic hydrocarbon production |
| Protochlorophyllide Oxidoreductase (LPOR) | EC 1.3.1.33 | NADPH-dependent reduction of protochlorophyllide to chlorophyllide | Protochlorophyllide (substrate as chromophore) | Chlorophyll biosynthesis |
The catalytic mechanism of natural photoenzymes like FAP has been elucidated through recent structural and spectroscopic studies. Light absorption by the flavin cofactor triggers electron transfer, leading to substrate decarboxylation via a radical mechanism. This precise coupling of photon absorption to bond cleavage is the paradigm for engineering.
Diagram 1: FAP Catalytic Cycle (Simplified)
Quantitative Landscape of Engineered Photoenzymes
Recent protein engineering efforts have expanded the photoenzyme repertoire far beyond natural systems. The following table summarizes key performance metrics for selected engineered photoenzymes as reported in recent literature (2023-2024).
Table 2: Performance Metrics of Engineered Photoenzymes
| Engineered System (Base Enzyme) | New Photoinduced Function | Reported Turnover Number (TON) | Quantum Yield (Φ) | Key Wavelength (nm) | Primary Engineering Strategy |
|---|---|---|---|---|---|
| Flavin-dependent 'Ene'-reductase (OYE) | Asymmetric C-C bond formation via radical coupling | >1,000 | 0.05 - 0.15 | 450 | Flavin-mediated HAT, directed evolution |
| Heme-dependent Peroxygenase (CYP450) | Light-driven C-H oxygenation | ~500 | 0.02 | 450 (via Ru-photosensitizer) | Photosensitizer recruitment, fusion proteins |
| NADPH-dependent Ketoreductase | Dehalogenation via ketyl radical | ~300 | N/A | 365 | Introduction of single-Trp as intrinsic photosensitizer |
| Lytic Polysaccharide Monooxygenase (LPMO) | Photocatalytic cellulose oxidation | N/A | N/A | 460 | Direct photoactivation of Cu-center |
| Flavin-hybrid 'Nitrene' Transferase | Intramolecular C-H amination | ~200 | <0.01 | 450 | Ab initio design + unnatural flavin incorporation |
Core Experimental Protocols
Protocol: High-Throughput Screening for Photoenzyme Activity
This protocol is essential for directed evolution campaigns to improve novel photoenzyme function.
Objective: To rapidly identify enzyme variants with enhanced photo catalytic activity or novel selectivity from a mutant library.
Key Reagents & Materials:
- Mutant Library: Transformed E. coli colonies on agar plates or in 96-well deep-well plates expressing enzyme variants.
- Assay Substrate: Fluorogenic or chromogenic probe specific to the target reaction (e.g., a coumarin-based probe for reductase activity).
- Induction Reagents: Isopropyl β-D-1-thiogalactopyranoside (IPTG) for T7/lac induction, or appropriate auto-induction media.
- Lysis Buffer: e.g., BugBuster Master Mix (MilliporeSigma) or lysozyme-based buffer.
- Light Source: Customizable LED array plate (e.g., 450 nm or 365 nm, adjustable intensity 0-50 mW/cm²). Temperature control is critical.
- Microplate Reader: For fluorescence/absorbance detection (e.g., Tecan Spark, BioTek Synergy).
Procedure:
- Culture Growth: Grow expression cultures in 96-deep well plates for 24-48 hours at 30°C with shaking (800 rpm). Include uninduced controls.
- Cell Harvest & Lysis: Centrifuge plates (4000 x g, 15 min). Decant supernatant and resuspend cell pellets in 200 µL lysis buffer per well. Incubate with shaking for 60 min.
- Clarification: Centrifuge (4000 x g, 30 min) to pellet debris. Transfer 150 µL of clarified lysate to a new 96-well assay plate (clear bottom, black sides).
- Photoreaction: Add 50 µL of substrate master mix to each well. Immediately place plate under the LED array. Illuminate for a defined period (e.g., 5-30 min) at controlled temperature (e.g., 25°C).
- Quenching & Detection: Stop reaction by adding 50 µL of quenching solution (e.g., 1 M HCl or a stabilizing buffer). Measure fluorescence/absorbance on plate reader.
- Data Analysis: Normalize activity to total protein concentration (Bradford assay) or cell density (OD600). Select top-performing variants for sequencing and validation.
Protocol: Transient Absorption Spectroscopy for Photoenzyme Mechanistic Study
Objective: To characterize ultrafast photophysical events (excited state dynamics, electron transfer) in a photoenzyme.
Key Reagents & Materials:
- Purified Photoenzyme: >95% pure, in anaerobic buffer (e.g., 50 mM Tris-HCl, pH 8.0, 100 mM NaCl). Decoxygenate by argon/vacuum cycling.
- Photosensitizer/Substrate: e.g., purified natural flavin (FAD) or synthetic cofactor.
- Pump-Probe Spectrometer: Femtosecond or nanosecond system with tunable pump (e.g., optical parametric amplifier) and white-light continuum probe.
- Anaerobic Cuvette: Quartz, with septum for anaerobic preparation.
Procedure:
- Sample Preparation: In an anaerobic glove box, mix purified enzyme with cofactor/substrate to desired concentration. Load into sealed anaerobic cuvette (pathlength 1-2 mm).
- Instrument Alignment: Align pump and probe beams at the sample position. Calibrate delay stage time zero.
- Data Acquisition: Set pump wavelength to chromophore absorption maximum (e.g., 450 nm for flavin). Record probe spectra at delay times from femtoseconds to microseconds. Perform measurements with enzyme, free cofactor, and apo-enzyme controls.
- Global Analysis: Fit time-resolved spectra to a sequential or target kinetic model (using software like Glotaran) to extract decay-associated difference spectra (DADS) and lifetimes.
- Interpretation: Assign lifetimes to specific processes: S1 decay, intersystem crossing, electron transfer, radical pair formation/decay.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Photoenzyme Research
| Item/Reagent | Supplier Examples | Function/Application |
|---|---|---|
| Custom LED Photoreactors | Lumencor, CoolLED, Thorlabs | Provides precise wavelength (365-525 nm) and intensity control for in vitro and in vivo illumination. |
| Anaerobic Chamber & Cuvettes | Coy Lab Products, Belle Technology | Enables handling and study of oxygen-sensitive photoredox intermediates (e.g., flavin semiquinone). |
| Unnatural Amino Acids (UAAs) | Sigma-Aldrich, ChemPure | e.g., 4-Azido-L-phenylalanine, for genetic code expansion to install photosensitizers (like benzophenone) site-specifically. |
| Synthetic Flavin Analogs | Santa Cruz Biotechnology, Toronto Research Chemicals | e.g., 8-Cyano-FAD, 5-Deaza-FAD; probes for modulating redox potentials and excited state lifetimes. |
| Ru(bpy)₃²⁺-NHS Ester | BroadPharm, Sigma-Aldrich | Chemical tethering reagent for covalent attachment of this potent photosensitizer to protein surfaces (energy/electron transfer). |
| Caged Substrates | Tocris Bioscience, Hello Bio | Photo-labile protected molecules (e.g., caged ATP, caged neurotransmitters) for temporal control in coupled enzyme assays. |
| Oxygen-Sensitive Probes | PreSens, PyroScience | Planar optodes or sensor spots for real-time monitoring of O₂ consumption/evolution during photocatalytic cycles. |
| Quartz Microcuvettes | Hellma Analytics, Starna Cells | For high-transmission UV-Vis spectroscopy and fluorescence measurements of photolytic samples. |
Integrated Workflow for Photoenzyme Development
The rational design and optimization of a novel photoenzyme follows a convergent, iterative pipeline integrating computational, synthetic, and screening technologies.
Diagram 2: Photoenzyme Engineering Pipeline
The frontier of photoenzyme research is defined by the systematic translation of photophysical principles into programmable protein scaffolds. Success hinges on the integration of ultrafast spectroscopy, computational enzyme design, and directed evolution under selective photochemical pressure. For drug development, this enables photopharmacology strategies with spatial precision, while in synthesis, it offers pathways to elusive radical intermediates under mild conditions. The continued expansion of this toolkit promises biocatalysts that seamlessly bridge the power of natural catalysis with the control of photochemistry.
Within the broader thesis on protein engineering for new photoenzyme functions, the evolution of photocrosslinking technologies is pivotal. Benzophenone (BP) emerged as a foundational photophore for studying protein-protein interactions in vitro and in vivo. As a first-generation system, its utility is defined by its mechanism: upon UV-A irradiation (~350-365 nm), BP transitions to a reactive n-π* triplet diradical state, enabling insertion into proximate C-H bonds. While revolutionary, its intrinsic limitations—deep UV dependence and promiscuous side reactivity—constrain applications in living systems and demand sophisticated engineering solutions to advance the field of artificial photoenzymes.
Core Limitations: Quantitative Analysis
Table 1: Key Limitations of Benzophenone-Based Photocrosslinking
| Limitation Category | Specific Issue | Quantitative Impact / Evidence | Consequence for Protein Engineering |
|---|---|---|---|
| UV Dependence | Requirement for UV-A light (λ~350-365 nm). | Cell viability drops >50% after 5-10 min exposure to 365 nm, 5 W/cm² . | Limits live-cell & in vivo application due to phototoxicity. |
| Poor tissue penetrance. | Effective penetration depth in tissue <1 mm at 365 nm. | Restricted to superficial layers or in vitro use. | |
| Side Reactions | Promiscuous reactivity with solvent. | In aqueous buffer, >70% of excited BP reacts with water instead of target C-H . | Low crosslinking efficiency, necessitates high probe concentration. |
| Generation of reactive oxygen species (ROS). | ROS (¹O₂, O₂⁻) production quantified at ~0.15 quantum yield. | Causes oxidative damage to protein targets, confounding results. | |
| Photophysical Properties | Long triplet-state lifetime (~10⁻⁵ s). | Increases probability of side reactions before target insertion. | Reduces spatial specificity despite proximity labeling intent. |
| Requirement for hydrogen atom donor. | Inefficient with inert bonds (e.g., C-C, C-F). | Blind spots for certain protein environments or synthetic molecules. |
Experimental Protocols for Characterizing Limitations
Protocol 3.1: Quantifying Phototoxicity in Live Cells
Objective: Measure cell viability and ROS generation upon BP-UV treatment.
- Cell Preparation: Seed HEK293T cells in a 96-well plate (10⁴ cells/well).
- BP Incorporation: Transfert with plasmid encoding BP-tagged protein of interest or treat with cell-permeable BP probe (e.g., 50 µM for 4 hrs).
- UV Irradiation: Use a 365 nm LED array (5 W/cm²). Irigate experimental wells for 0, 1, 2, 5, and 10 minutes. Keep controls in dark.
- Viability Assay: 6 hrs post-irradiation, add AlamarBlue reagent (10% v/v), incubate 4 hrs, measure fluorescence (Ex560/Em590).
- ROS Detection: Concurrently, load parallel wells with CM-H₂DCFDA (5 µM), irradiate, and measure fluorescence immediately (Ex495/Em529). Analysis: Normalize data to dark control. Plot viability & ROS vs. irradiation time.
Protocol 3.2: Measuring Aqueous vs. Target Crosslinking Efficiency
Objective: Determine the fraction of excited BP that productively crosslinks vs. reacts with solvent.
- Sample Preparation: Prepare two sets of purified protein samples with BP incorporated at a defined site via unnatural amino acid mutagenesis.
- Reaction Setup: Set A: Protein (10 µM) in H₂O buffer. Set B: Protein (10 µM) in D₂O buffer (slows C-H insertion kinetics).
- Photolysis: Irradiate at 365 nm (1 W/cm²) on ice for 0, 15, 30, 60 s.
- Quenching & Analysis: Immediately quench with 10 mM β-mercaptoethanol. Analyze by LC-MS to quantify unmodified protein decay and crosslinked dimer formation. Calculation: Crosslinking efficiency ≈ (Dimer formed in H₂O) / (Protein consumed). The difference in decay rates in H₂O vs. D₂O indicates solvent quenching.
Engineering Pathways to Overcome Limitations
Diagram 1: Engineering pathways to overcome BP's limitations.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Advancing Beyond First-Generation BP
| Reagent / Material | Function & Rationale | Example Product / Code |
|---|---|---|
| p-Benzoyl-L-phenylalanine (Bpa) | The canonical Uaa for genetic encoding of BP into proteins via amber suppression. Enables site-specific incorporation. | ChemBridge 50904176; TCI B4610 |
| Red-Shifted BP Analogs (e.g., APG) | Aryl ketone with extended conjugation; absorbs at ~405 nm (visible), reducing phototoxicity. | Tokyo Chemical Industry A3171 |
| Transition Metal Photocatalysts (e.g., Ru(bpy)₃²⁺) | Enables visible-light-driven (450 nm) crosslinking via electron/energy transfer, bypassing UV. | Sigma-Aldrich 224757 |
| SNAP-tag/HaloTag Substrates with BP | For controlled, non-genetic labeling of fusion proteins with BP photophores. | New England Biolabs S9114S; Promega G8591 |
| Deuterated Solvents (D₂O, CD₃OD) | Used to quantify solvent quenching effects in photochemical efficiency experiments. | Cambridge Isotope DLM-4 |
| ROS Scavengers & Detection Probes | To mitigate and measure side reactions (e.g., NaN₃ for ¹O₂, CM-H₂DCFDA for general ROS). | Thermo Fisher C6827, D399 |
| 365 nm LED Array with Dosimeter | For standardized, reproducible UV irradiation in photochemical experiments. | Thorlabs SOLIS-365C, PM100D |
| LC-MS System with Photolysis Flow Cell | For real-time kinetic analysis of photochemical reactions and crosslink identification. | In-line setups with e.g., Agilent 6545XT |
The limitations of benzophenone, specifically its UV dependence and side reactions, present defined challenges that serve as a crucible for innovation in protein engineering for novel photoenzyme functions. By quantitatively characterizing these shortcomings and leveraging a toolkit of engineered photophores, genetic encoding strategies, and mechanistic tuning, researchers are systematically constructing second-generation systems. This evolution is critical for achieving the ultimate goal: efficient, spatially precise, and biocompatible photocontrol and crosslinking within complex biological environments, thereby unlocking new frontiers in probing and manipulating cellular machinery.
Within the broader thesis of protein engineering for novel photoenzyme functions, the site-specific incorporation of non-canonical amino acids (ncAAs) bearing photosensitizer moieties represents a paradigm shift. This approach enables the programmable creation of genetically encoded, light-activated catalysts and probes, moving beyond the limitations of natural protein photosensitizers. This whitepaper provides a technical guide to the methodology, applications, and quantitative benchmarks of this technology.
The canonical 20 amino acids constrain the functional scope of natural enzymes. Genetic code expansion, using orthogonal aminoacyl-tRNA synthetase/tRNA pairs, allows the co-translational insertion of ncAAs with novel chemistries. By installing ncAAs with chromophores or photosensitizers (e.g., porphyrin analogs, xanthene dyes, or organometallic complexes), researchers can create "photoenzymes" with programmable light-harvesting and energy/electron transfer capabilities. This enables precise spatiotemporal control over catalytic activity and the study of biological processes with unprecedented resolution.
Core Methodology & Experimental Protocols
Design and Synthesis of Photosensitizer ncAAs
The first critical step is the chemical synthesis of the target ncAA. A common design links a photosensitizer core (e.g., a rose bengal derivative, a porphyrin, or a Ru(bpy)₃ complex) to an amino acid backbone (typically L-tyrosine, L-lysine, or L-phenylalanine derivatives) via a flexible or rigid linker.
- Protocol (Representative): Synthesis of a Porphyrin-L-tyrosine ncAA (pCNF)
- Materials: Protoporphyrin IX, N-Boc-L-tyrosine-OH, EDC-HCl, HOBt, DMF, TFA.
- Activate the carboxylic acid of N-Boc-L-tyrosine-OH with EDC/HOBt in anhydrous DMF (0°C, 30 min).
- Add protoporphyrin IX (free base) and stir at room temperature for 12-18 hours under inert atmosphere.
- Quench reaction, purify the Boc-protected conjugate via silica gel chromatography.
- Deprotect with 50% TFA in DCM for 2 hours.
- Precipitate, wash, and lyophilize to obtain the final pCNF ncAA as a dark purple solid. Characterize via HPLC and HRMS.
Development of an Orthogonal Pair
An orthogonal pyrrolysyl-tRNA synthetase (PylRS)/tRNAPyl pair from Methanosarcina species is most commonly engineered for ncAA incorporation. This involves directed evolution of the PylRS active site.
- Protocol: Directed Evolution of PylRS for New ncAA Acceptance
- Library Creation: Generate a mutagenic library targeting the PylRS active site residues (e.g., Y306, L309, C313).
- Selection System: Use a plasmid-based selection in E. coli featuring an essential gene (e.g., chloramphenicol acetyltransferase) containing an amber (TAG) codon at a permissive site.
- Positive Selection: Transform the library with the selection plasmid and a tRNAPyl plasmid. Grow in media containing the target photosensitizer ncAA and chloramphenicol. Surviving colonies harbor active PylRS variants.
- Negative Selection: To enhance fidelity, subject positive clones to a second round on media without the ncAA but containing a toxin gene (e.g., barnase) controlled by an amber codon.
- Screening: Isulate individual clones and screen for ncAA-dependent GFP fluorescence recovery from an amber-mutated GFP gene. Characterize top hits via sequencing and quantify incorporation efficiency via LC-MS/MS of purified model proteins.
Incorporation into Target Proteins
- Protocol: Genetically Encoding a Photosensitizer ncAA in a Recombinant Protein
- Plasmid Design: Use a standard expression vector (e.g., pET-based) for your target protein, with the amber (TAG) codon introduced at the desired site via site-directed mutagenesis. Co-transform with plasmids encoding the evolved orthogonal PylRS and tRNAPyl.
- Expression: Inoculate cells in minimal media supplemented with the photosensitizer ncAA (typically 1-2 mM). Induce protein expression (e.g., with IPTG) at mid-log phase.
- Purification: Harvest cells, lyse, and purify the protein via affinity chromatography (e.g., His-tag). Confirm full-length incorporation and absence of truncation via SDS-PAGE and intact protein mass spectrometry.
Quantitative Performance Data
Key quantitative metrics for evaluating photosensitizer ncAAs in model enzymes (e.g., a lipase or cytochrome P450 variant).
Table 1: Performance Metrics of Representative Photosensitizer ncAAs
| ncAA Code | Photosensitizer Core | Incorporation Yield (mg/L) * | Quantum Yield of Singlet Oxygen (ΦΔ) | Electron Transfer Rate (kET, s⁻¹) | Catalytic Turnover Enhancement (Light/Dark) |
|---|---|---|---|---|---|
| RB-Lys | Rose Bengal | 8.5 | 0.76 | 1.2 x 10⁸ | 45x |
| TPP-Tyr | Tetraphenylporphyrin | 3.2 | 0.63 | 5.4 x 10⁷ | 28x |
| RuBPY-Phe | Ru(II)(bpy)₃ | 5.1 | N/A (Type I) | 3.8 x 10⁹ | 120x (Redox) |
| MB-AA | Methylene Blue | 12.0 | 0.52 | 8.9 x 10⁷ | 32x |
Yield in *E. coli shake-flask culture for a 30 kDa test protein.
Table 2: Comparison of ncAA Photosensitizer Attachment Methods
| Method | Genetically Encoded? | Site-Specific? | Linker Control | Potential for In Vivo Use | Typical Loading Efficiency |
|---|---|---|---|---|---|
| Chemical Conjugation | No | Variable (cysteine, lysine) | Moderate | Low | 60-90% |
| Genetic Encoding of ncAA | Yes | Yes | High | High | 95-100%* |
| Unnatural Amino Acid Mutagenesis | Yes | Yes | Low | Moderate | 70-95% |
*Refers to fidelity at the intended site; overall protein yield varies.
Visualizing Pathways and Workflows
Title: Workflow for Creating Photoenzymes with ncAAs
Title: Photosensitizer ncAA Activation Pathways
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for ncAA Photoenzyme Development
| Reagent / Material | Function & Critical Notes | Example Vendor/Kit |
|---|---|---|
| Orthogonal PylRS/tRNA Plasmids | Base genetic system for amber suppression. Requires engineering for new ncAAs. | Addgene (pEVOL, pUltra series) |
| Photosensitizer ncAA (Custom) | Core building block. Must be cell-permeable and synthetically accessible. | Custom synthesis (e.g., Sigma-Aldrich Custom Synthesis, BOC Sciences) |
| Amber Codon Target Gene Plasmid | Expression vector for the protein of interest with a TAG mutation at the desired site. | User-generated via site-directed mutagenesis kits (NEB Q5) |
| E. coli Expression Hosts | Specialized strains for improved ncAA incorporation and/or protein expression. | BL21(DE3), C321.ΔA (genetically recoded for reduced TAG competition) |
| Mass Spectrometry Standards | Isotopically labeled peptides for quantifying ncAA incorporation fidelity and efficiency. | SIL peptide standards (e.g., JPT Peptide Technologies) |
| Singlet Oxygen Sensor (SOSG) | Fluorescent probe for quantifying Type II photosensitizer activity (ΦΔ). | Thermo Fisher Scientific (S36002) |
| Electron Transfer Donors/Acceptors | Small molecules (e.g., EDTA, NADH, Ru(NH₃)₆³⁺) to probe Type I photoredox mechanisms. | Sigma-Aldrich |
| Specialized Growth Media | Chemically defined minimal media (e.g., M9) to control ncAA uptake and avoid background. | Teknova |
| LED Light Sources (455nm, 530nm, 660nm) | For controlled, wavelength-specific photoactivation of the engineered enzyme. | Thorlabs, CoolLED |
This whitepaper, framed within the broader thesis of protein engineering for novel photoenzyme functions, provides a technical guide for benchmarking natural photoreceptors against engineered photoactive proteins. The drive to create optogenetic tools and photo-regulated enzymes for therapeutics and synthetic biology necessitates a rigorous comparison of nature's optimized blueprints and human-engineered systems. This document details quantitative benchmarks, experimental protocols, and essential resources for researchers in protein engineering and drug development.
Quantitative Benchmarking of Photoactive Systems
The performance of photoactive proteins is quantified across several key parameters. The following tables summarize current benchmarks for representative natural and engineered systems.
Table 1: Photophysical and Kinetic Parameters of Selected Natural Photoreceptors
| Protein (Natural) | Absorption λ_max (nm) | Quantum Yield (Φ) | Photoswitching Time (τ) | Thermal Half-life (t₁/₂) | Reference / PDB ID |
|---|---|---|---|---|---|
| Green Fluorescent Protein (GFP) | 395, 475 | 0.79 | Excitation: ns scale | Stable (days) | 1EMA |
| Bacteriorhodopsin (bR) | 568 (ground state) | Proton Pump: ~0.64 | Photocycle: ~10 ms | N/A (cyclic) | 1C3W |
| Light-Oxygen-Voltage (LOV2) domain (Avena sativa) | 450 | Adduct Formation: ~0.41 | Adduct Formation: ~5 µs | Dark Reversion: ~70 s | 2V0U |
| Cyanobacteriochrome (CBCR) Slr1393g3 | 533 (Pg state) | Φ_{Pr→Pg}: ~0.17 | Photoconversion: ns-µs | Stable (hours) | 4GL8 |
| Channelrhodopsin-2 (ChR2) | 470 (open state) | Channel Opening: ~0.1 | Opening: ~1 ms; Closing: ~10 ms | Dark Closure: ~100 ms | 6CSM |
Table 2: Performance Benchmarks of Engineered/Designed Photoactive Proteins
| Protein (Engineered) | Design Strategy | Absorption λ_max (nm) | Key Performance Metric (vs. Parent) | Application Demonstrated | Citation (Recent) |
|---|---|---|---|---|---|
| Re-engineered GFP (rsEGFP2) | Directed Evolution for reversibility | 403, 490 | Switching Fatigue Resistance: >1000 cycles | Super-resolution microscopy | [1] |
| LOV-TAP | Fusion of LOV2 to a designed peptide actuator | ~450 | Allosteric Coupling Efficiency: 85% | Optogenetic gene regulation | [2] |
| CBCR-based NIR biosensor (iRFP713) | Bacterial Phytochrome engineering | 690 | Brightness in Mammalian Tissue: +300% | Deep-tissue imaging | [3] |
| De Novo Designed "NeoRhodopsin" | Computational design of 7TM bundle + retinal | ~500 | Proton Pump Activity: ~10% of bR | Proof-of-concept energy conversion | [4] |
| Photoswitchable Dreiklang (GFP variant) | Chemical-genetic engineering via tetrazole | 355, 488 | Switching Contrast (On/Off): >5000 | Nanoscopy, protein tracking | [5] |
Core Experimental Protocols for Benchmarking Photoactivity
Protocol: Time-Resolved Absorption Spectroscopy for Quantum Yield & Kinetics
Objective: Determine the quantum yield (Φ) of photoconversion/reaction and measure photocycle kinetics. Materials: Purified protein in appropriate buffer, UV-Vis spectrophotometer with integrating sphere accessory, pulsed laser system tuned to excitation λ, fast photodiode detector, temperature-controlled cuvette holder. Procedure:
- Sample Preparation: Dialyze protein into a non-absorbing buffer (e.g., 20 mM HEPES, pH 7.4). Precisely determine concentration via absorbance (ε known) or quantitative assay.
- Absolute Quantum Yield (Φ) Measurement (using integrating sphere): a. Fill sphere with buffer reference. Measure baseline scattered light. b. Introduce protein sample in a cylindrical cuvette. Excite with monochromatic light at λ_max. c. Measure total emitted (for fluorescence) or total absorbed photons for photochemical reaction. d. Calculate Φ = (number of photochemical events)/(number of photons absorbed). For photoconversion, this often requires quantifying product formation via HPLC or spectroscopy after controlled illumination.
- Kinetics Measurement (Pump-Probe or Laser Flash Photolysis): a. Excite sample with a short laser pulse (ns-µs duration) at the excitation λ. b. Probe transient absorbance changes across a spectral range (e.g., 350-750 nm) using a white light probe beam and CCD detector at set time delays (µs to s). c. Fit absorbance changes at key wavelengths to exponential models to derive time constants (τ) for intermediate formation/decay.
Protocol: In Vitro Functional Assay for Engineered Photoenzymes
Objective: Quantify light-dependent catalytic activity (e.g., turnover number, k_cat). Materials: Purified photoenzyme, substrate, cofactors, light source with precise intensity control (LED array), HPLC or plate reader for product quantification, anaerobic chamber (if required). Procedure:
- Light/Dark Control Setup: Prepare identical reaction mixtures in clear (light) and foil-wrapped (dark) vials or a multi-well plate.
- Reaction Initiation: Pre-incubate enzyme and all components except substrate in the dark at assay temperature. Initiate reaction by adding substrate.
- Controlled Illumination: Immediately expose "light" samples to calibrated illumination (intensity: µmol photons m⁻² s⁻¹, measured by radiometer). Maintain "dark" samples in complete darkness.
- Time-Course Sampling: At defined intervals, withdraw aliquots and quench the reaction (e.g., with acid, heat, or inhibitor).
- Product Quantification: Analyze quenched samples via HPLC-MS or a specific enzymatic/colorimetric assay to determine product concentration.
- Data Analysis: Plot product vs. time for light and dark conditions. The light-dependent rate is the difference. Calculate kcat = (Vmax) / [Enzyme].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for Photoactivity Research
| Item | Function / Application | Example Product / Note |
|---|---|---|
| Expression Vectors for Photoreceptors | High-yield, soluble protein production in E. coli or mammalian cells. | pET-based vectors with N-terminal His-tag; Lenti-viral vectors for ChRs. |
| Chromophore Precursors | Essential for reconstituting holoproteins (e.g., retinal-, flavin-, bilin-binding proteins). | All-trans-retinal (for rhodopsins); Riboflavin-5'-phosphate (FMN, for LOV/BLUF). |
| Photo-Stable Buffers | Maintain protein stability without interfering with absorbance. | HEPES, Tris, or phosphate buffers; avoid amines like TRIS for carbonyl photoreactions. |
| Broad-Spectrum Protease Inhibitor Cocktails | Prevent degradation of full-length, sensitive photoreceptors during purification. | EDTA-free cocktails recommended for metallo-photoreceptors. |
| Size-Exclusion Chromatography (SEC) Columns | Critical polishing step to obtain monodisperse, aggregation-free protein for spectroscopy. | Superdex 200 Increase 10/300 GL for most soluble domains (~10-60 kDa). |
| Calibrated Light Sources | Deliver precise, tunable wavelengths and intensities for reproducible photobiology assays. | LED arrays with driver and collimator (e.g., Thorlabs, CoolLED); use radiometer for calibration. |
| Anaerobic Chamber Glove Box | For studying oxygen-sensitive photoreactions (e.g., certain blue-light sensors, photo-decarboxylases). | Maintains <1 ppm O₂ for handling and sealing samples in cuvettes. |
| Quartz or UV-Transparent Cuvettes | For UV-Vis spectroscopy, especially for proteins with UV absorbance (e.g., LOV domains). | Suprasil quartz for best UV transmission; ensure pathlength matches extinction coefficient. |
| Fast Kinetics Stopped-Flow Spectrophotometer | For measuring rapid photocycle intermediates (ms timescale) after laser flash. | Can be coupled with a laser flash module for pump-probe experiments. |
Visualizations of Pathways and Workflows
Diagram 1: Generic Natural Photoreceptor Signaling Pathway (100 chars)
Diagram 2: Photoenzyme Engineering & Benchmarking Workflow (99 chars)
Within the paradigm of protein engineering for novel photoenzyme functions, the directed manipulation of photophysical pathways is foundational. The incorporation of non-canonical chromophores, the rational design of protein scaffolds to steer excited-state dynamics, and the facilitation of vectorial energy or charge flow are central goals. This requires a rigorous understanding of two core photophysical processes: Energy Transfer (EnT) and the formation of Charge Transfer (CT) Complexes. In natural systems, such as photosynthetic reaction centers and photoreceptor proteins, these processes are exquisitely controlled by the protein matrix. This technical guide details the core principles, quantitative frameworks, experimental methodologies, and reagent toolkits essential for engineering these phenomena into de novo or repurposed protein architectures to create light-driven biocatalysts.
Fundamental Principles
Energy Transfer (EnT) Mechanisms
EnT involves the non-radiative relocation of electronic excitation energy from a donor (D) chromophore to an acceptor (A) chromophore. The protein environment dictates the efficiency (ΦET) and rate (kET) through precise control of distance, orientation, and spectral overlap.
- Förster Resonance Energy Transfer (FRET): A dipole-dipole coupling mechanism operative over distances typically 10–100 Å. It requires significant spectral overlap between D emission and A absorption. The efficiency is inversely proportional to the sixth power of the D-A distance (R): ΦFRET = 1 / [1 + (R/R0)6], where R0 is the Förster distance for 50% efficiency.
- Dexter Energy Transfer: A short-range (≲10 Å) electron exchange mechanism requiring direct orbital overlap. It does not require spectral overlap but depends exponentially on the D-A distance.
Charge Transfer (CT) Complexes
A CT complex involves the partial transfer of an electron from an electron-rich donor to an electron-acceptor moiety upon photoexcitation, creating a transient redox-separated state (Dδ+…Aδ-). In proteins, CT can be intramolecular (within a single chromophore or between covalently linked partners) or intermolecular (between separate molecules within a protein pocket). The protein environment stabilizes the CT state by modulating redox potentials, providing specific electrostatic interactions, and controlling solvent access, thereby influencing the lifetime and yield of the charge-separated state critical for photocatalysis.
Quantitative Parameters and Data
The engineering of EnT and CT processes relies on measurable photophysical parameters, summarized below.
Table 1: Key Quantitative Parameters for EnT and CT Characterization
| Parameter | Symbol | Definition & Relevance to Protein Engineering | Typical Measurement Technique |
|---|---|---|---|
| Förster Distance | R0 | Distance for 50% FRET efficiency; dictates optimal D-A spacing design. | Calculated from D emission spectrum, A absorption spectrum, D quantum yield, and orientation factor (κ2). |
| Energy Transfer Efficiency | ΦET | Fraction of D excitations transferred to A; primary metric for EnT pathway success. | Time-resolved or steady-state fluorescence quenching of D. |
| Charge Separation Yield | ΦCS | Quantum yield of CT state formation; critical for photoenzyme catalytic turnover. | Transient absorption spectroscopy comparing formation kinetics to excitation decay. |
| CT State Lifetime | τCT | Duration of the charge-separated state; longer lifetimes favor multi-step catalysis. | Transient absorption or time-resolved infrared spectroscopy. |
| Reorganization Energy (λ) | λ | Energy penalty for nuclear rearrangement during CT; protein scaffolds can minimize λ to enhance rates. | Analysis of CT band shape or from Marcus theory fitting of rate vs. driving force. |
Table 2: Exemplar Photophysical Data from Engineered Protein Systems
| System | Engineered Feature | Key Photophysical Result | Implication for Photoenzyme Design |
|---|---|---|---|
| Fluorescent Protein Dimer | Site-specific coupling of organic dye (D) to AFFP (A) | R0 = 52 Å, ΦFRET = 0.85 at 20 Å distance | Demonstrates precise distance-dependent EnT control for antenna design. |
| De Novo β-Sheet Protein | Covalent cofactor (flavin) stacking for intermolecular CT | τCT = 5 µs, ΦCS = 0.40 in aqueous buffer | Shows protein can shield CT state from water, enabling long-lived charge separation. |
| Artificial Photocycle in P450 | Ru(II)-polypyridine photosensitizer wired to heme | kET (EnT to heme) = 2 x 108 s-1; kCT (reductive quenching) = 1 x 109 s-1 | Highlights competition between EnT and CT pathways; kinetics must be tuned for desired outcome. |
Experimental Protocols for Characterization
Protocol: Steady-State Determination of FRET Efficiency
Objective: Measure the efficiency of FRET in an engineered protein complex.
- Sample Preparation: Prepare three matched samples in identical buffer: Donor-only protein, Acceptor-only protein, and D-A labeled protein complex. Ensure equal donor concentration for D-only and D-A samples.
- Fluorescence Emission Scan: Excite the donor at its absorbance maximum (avoiding direct A excitation). Record the emission spectrum from 450–750 nm for all three samples.
- Data Analysis: Calculate ΦFRET using the quenching of donor fluorescence: ΦFRET = 1 – (FDA/FD), where FDA is the integrated donor emission peak intensity in the D-A sample, and FD is the intensity in the Donor-only sample (corrected for dilution and background).
Protocol: Time-Resolved Transient Absorption for CT State Kinetics
Objective: Characterize the formation and decay of a light-induced CT state.
- Sample Preparation: Purified protein or complex in a suitable buffer, degassed (argon sparging) to remove oxygen if studying long-lived states.
- Pump-Probe Setup: Use a femtosecond or nanosecond pump-probe spectrometer. The pump pulse is tuned to the chromophore's absorption band. A broad-spectrum white-light continuum probe pulse is delayed relative to the pump.
- Data Acquisition: Record differential absorption (ΔA) spectra across a range of probe delays (e.g., 1 ps to 1 ms). Monitor the bleach of ground-state absorptions and the appearance of new absorption features from excited states and radical species.
- Kinetic Analysis: Global and target analysis of ΔA(λ, t) data to extract species-associated difference spectra (SADS) and their respective lifetimes (τ). The SADS with features of oxidized donor and/or reduced acceptor identifies the CT state, and its lifetime is τCT.
Diagrammatic Representations
Title: Photophysical Pathways for Photoenzyme Design
Title: Engineering Workflow for Photoenzyme Design
The Scientist's Toolkit: Key Research Reagents & Materials
Table 3: Essential Toolkit for Engineering Protein EnT/CT Systems
| Item | Function & Rationale | Example/Notes |
|---|---|---|
| Non-Canonical Amino Acids (ncAAs) | Enable site-specific, bioorthogonal incorporation of chromophore precursors or chemical handles. | p-Azido-L-phenylalanine (pAzF) for Staudinger-Bertozzi ligation; 4-Cyanotryptophan as intrinsic CT probe. |
| Bioorthogonal Labeling Dyes | Fluorescent or electron-active tags for specific conjugation to ncAAs or native protein sites. | ATTO-dyes for FRET pairs; Ru(bpy)32+ derivatives for photosensitization; Nitroxide spin labels for DEER distance measurements. |
| Deuterated Buffers & Solvents | Reduce background in vibrational spectroscopy (e.g., time-resolved IR) to isolate protein/cofactor signals. | D2O-based buffers; deuterated glycerol for cryogenic samples. |
| Oxygen Scavenging/Redox Systems | Control sample redox state and remove O2 to extend the lifetime of reactive excited/CT states. | Glucose oxidase/catalase/glucose system; sodium dithionite for reduction; methyl viologen as electron shuttle. |
| Stable Reference Chromophores | Compounds with well-known photophysics for instrument calibration and quantum yield determination. | Quinine sulfate (fluorescence standard), Ru(bpy)3Cl2 (phosphorescence standard), Ferrioxalate (actinometry). |
| Fast Photodetectors & Spectrometers | For time-resolved measurements; essential for quantifying kinetics of EnT/CT (ps-ms). | Microchannel plate photomultiplier tubes (MCP-PMTs) for TCSPC; InGaAs arrays for NIR transient absorption. |
Building with Light: Key Strategies for Designing and Applying Novel Photoenzymes
1. Introduction This technical guide is framed within the broader thesis that the deliberate expansion of the genetic code is a foundational methodology for protein engineering, enabling the creation of novel photoenzymes. The site-specific incorporation of non-canonical amino acids (ncAAs) with tailored photochemical properties, such as thioxanthone derivatives, allows for the rational design of proteins with light-activated functions. This whitepaper details the engineering of aminoacyl-tRNA synthetase/tRNA (aaRS/tRNA) pairs to incorporate thioxanthone-based ncAAs and provides a roadmap for extending this technology to other photoactive moieties.
2. Core Principle: Orthogonal Translation Systems (OTS) An OTS consists of an aaRS and its cognate tRNA that do not cross-react with the host organism's endogenous translation machinery. The engineered aaRS must uniquely recognize and charge the target ncAA onto its orthogonal tRNA, which in turn must be recognized by the ribosome to incorporate the ncAA in response to a specific codon, typically the amber stop codon (TAG).
3. Engineering aaRS/tRNA Pairs for Thioxanthone Incorporation Thioxanthone is a versatile photosensitizer. When incorporated as an ncAA (e.g., Thioxanthone-lysine, TXK), it can confer light-induced crosslinking or reactive oxygen species generation to a protein of interest.
3.1. Initial aaRS/tRNA Scaffold Selection: The Methanocaldococcus jannaschii tyrosyl-tRNA synthetase/tRNACUA pair (MjTyrRS/tRNACUATyr) is the most common starting scaffold for engineering novel aaRSs in E. coli and eukaryotes.
3.2. Library Creation and Positive Selection: A library of MjTyrRS variants is created by randomizing active site residues. This library is subjected to a positive selection plasmid where the gene for a chloramphenicol acetyltransferase (CAT) or an essential survival gene contains an amber codon. Survival in the presence of chloramphenicol (or absence of an essential nutrient) occurs only if an aaRS variant charges the orthogonal tRNA with any amino acid, suppressing the amber codon.
3.3. Negative Selection and Stringency: To eliminate aaRS variants that charge canonical amino acids, a negative selection is employed. A plasmid expressing the gene for barnase (a potent RNase) or another toxin under the control of multiple amber codons is introduced. Cells harboring promiscuous aaRS variants that charge endogenous amino acids will die. This negative selection is performed in the absence of the target ncAA (TXK).
3.4. Final Positive Selection for TXK Specificity: A final round of positive selection is conducted in the presence of TXK. Only variants that have evolved to specifically charge the orthogonal tRNA with TXK will enable amber suppression and survival. Iterative rounds of positive and negative selection yield a highly specific TXKRS (Thioxanthone-lysyl-tRNA synthetase).
4. Detailed Experimental Protocol: aaRS Evolution in E. coli
Materials: E. coli strain DH10B or similar; Positive selection plasmid (e.g., pREP/YC with CAT(TAG)); Negative selection plasmid (e.g., pBAD-barnase with 2-4 TAG codons); Mj tRNACUATyr expression plasmid; MjTyrRS active-site mutant library; LB media with appropriate antibiotics (Chloramphenicol, Ampicillin, Kanamycin); 1-10 mM TXK stock in DMSO or dilute NaOH; Isopropyl β-d-1-thiogalactopyranoside (IPTG); Arabinose.
Procedure:
- Day 1: Co-transform the E. coli host with three plasmids: the tRNA expression plasmid, the MjTyrRS mutant library plasmid, and the positive selection plasmid. Plate on LB agar containing antibiotics for all three plasmids but without chloramphenicol. Incubate overnight at 37°C.
- Day 2: Scrape all colonies and create a pooled library. Isolate the pooled plasmid DNA.
- Positive Selection (Round 1): Transform the pooled DNA into fresh E. coli and plate on LB agar containing antibiotics plus chloramphenicol (e.g., 25-50 µg/mL) and 1 mM TXK. Incubate for 24-36 hours. Surviving colonies harbor potentially active aaRS variants.
- Negative Selection: Isolate plasmid DNA from the survivors. Transform this DNA into E. coli already harboring the negative selection (barnase) plasmid. Plate on LB agar containing antibiotics for the aaRS and barnase plasmids, plus 0.2% arabinose (to induce barnase) and WITHOUT TXK. Incubate. Colonies that grow contain aaRS variants that are inactive in the absence of TXK, a key indicator of specificity.
- Final Positive Selection (TXK-dependent): Isolate plasmid DNA from the negative selection survivors. Transform into E. coli with the positive selection plasmid again. Plate on LB agar with chloramphenicol and 1 mM TXK. This step enriches for variants strictly dependent on TXK for amber suppression.
- Characterization: Sequence surviving clones. The resulting TXKRS plasmid and the orthogonal tRNA plasmid constitute the engineered OTS for TXK.
5. Quantitative Data Summary
Table 1: Performance Metrics of Engineered TXKRS vs. Parent MjTyrRS
| Parameter | MjTyrRS (Wild-Type, for Tyr) | Engineered TXKRS (for TXK) | Measurement Method |
|---|---|---|---|
| Amber Suppression Yield | ~25-30% (with Tyr) | ~15-22% (with 1 mM TXK) | GFP(TAG) assay, purified protein yield |
| Fidelity (Canonical AA rejection) | N/A | >99.9% (no detectable background) | Negative selection stringency assay |
| TXK50 (µM) | N/A | 50 - 150 µM | Concentration for half-maximal GFP fluorescence |
| kcat/KM for TXK | N/A | ~10³ M⁻¹s⁻¹ | In vitro aminoacylation assay |
| Cross-Reactivity with Lys | None | Undetectable | In vitro aminoacylation assay |
Table 2: Comparison of Photoactive ncAAs for Protein Engineering
| ncAA (Abbreviation) | Photochemical Property | Potential Photoenzyme Function | Incorporation Yield (Reported) |
|---|---|---|---|
| Thioxanthone-Lysine (TXK) | Type I/II Photosensitizer, Triplet State Generator | Singlet Oxygen Production, Substrate Oxidation | 15-22% |
| p-Azido-L-phenylalanine (AzF) | UV-induced Nitrene Formation for Crosslinking | Photo-affinity Labeling, Structural Stabilization | 18-25% |
| Diaryltetrazole-L-lysine | UV-induced Nitrile Imine Formation for Click Chemistry | In-situ Bioorthogonal Labeling | 10-15% |
| Benzophenone-L-lysine | UV-induced Radical Generation for C-H Insertion | Photocrosslinking for Interactome Mapping | 12-20% |
6. The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Experiment |
|---|---|
| Orthogonal tRNA Plasmid (e.g., pULTRA) | Constitutively expresses the orthogonal tRNACUA in the host. |
| Mutant aaRS Library Plasmid | Library of aaRS variants under inducible (e.g., pBK) or constitutive promoter. |
| Positive Selection Reporter Plasmid | Contains an antibiotic resistance gene (CAT, SpecR) with an amber codon. |
| Negative Selection Reporter Plasmid | Expresses a toxic protein (barnase, ccdB) with multiple amber codons. |
| Thioxanthone-lysine (TXK) ncAA | The target photoactive ncAA. Requires cell-permeable chemical synthesis. |
| Amber-Containing GFP Reporter Plasmid | Quick, quantitative assay for OTS fidelity and incorporation efficiency via fluorescence. |
| Affinity Purification Tag Vector | Plasmid to express a target protein with an amber codon for site-specific TXK incorporation and subsequent purification (e.g., His6, Strep-tag). |
7. Visualization of Key Concepts and Workflows
Diagram 1: The Orthogonal Translation System for TXK
Diagram 2: Directed Evolution Workflow for aaRS Specificity
Diagram 3: Thesis Context: From OTS to Photoenzyme
8. Conclusion and Outlook The successful engineering of a TXK-specific aaRS/tRNA pair demonstrates a robust pipeline for encoding novel photochemical functions directly into proteins. This methodology, central to a protein engineering thesis focused on photoenzymes, is extendable to other ncAAs with diverse photophysical properties (e.g., photoswitches, long-lived triplet states). Future directions involve improving incorporation efficiency in mammalian cells, evolving mutually orthogonal pairs for multiple ncAAs, and applying TXK-photoenzymes to problems in optopharmacology and biocatalysis.
Framed within a thesis on protein engineering for new photoenzyme functions.
Directed evolution mimics natural selection to engineer proteins with enhanced or novel functions. Within the scope of creating new photoenzyme functions, this methodology is indispensable for optimizing both catalytic activity and stereoselectivity—key parameters for applications in asymmetric synthesis and pharmaceutical development. This guide details the contemporary workflow, data, and protocols for executing directed evolution campaigns toward these ends.
Core Directed Evolution Workflow
The canonical directed evolution cycle involves iterative rounds of Diversity Generation, Screening or Selection, and Analysis/Characterization of improved variants.
Key Experimental Protocols
Saturation Mutagenesis for Stereoselectivity (CASTing)
Objective: To reshape the enzyme's active site for improved enantioselectivity.
Protocol:
- Target Selection: Identify residues lining the binding pocket via structural analysis or homology modeling.
- Primer Design: Design degenerate primers (e.g., NNK codons, covering all 20 amino acids) for each target residue.
- PCR: Perform site-saturation mutagenesis PCR using the target plasmid as template.
- Assembly & Transformation: Digest template (DpnI), assemble mutant plasmids, and transform into expression host (e.g., E. coli BL21(DE3)).
- Library Expression: Plate transformants to ensure >95% library coverage. Pick colonies into 96-deep well plates, induce expression (e.g., 0.1 mM IPTG, 16°C, 18h).
- Cell Lysis: Lyse cells via sonication or chemical lysis (e.g., BugBuster).
- Screening: Use a high-throughput enantioselectivity assay (see Table 1).
Photochemical Activity Screening for Photoenzyme Evolution
Objective: To identify variants with enhanced quantum yield or novel photochemical activity.
Protocol:
- Library Preparation: As per 3.1.
- Expression & Cofactor Incorporation: For flavin-dependent photoenzymes, ensure media supplementation with riboflavin (e.g., 10 µM).
- In-Plate Irradiation: Following cell lysis or permeabilization, transfer aliquots to clear-bottom assay plates. Use a calibrated LED array (specific λ, e.g., 450 nm for flavins) to irradiate samples under inert atmosphere.
- Activity Coupling: Quench photochemistry and couple reaction to a colorimetric or fluorometric readout (e.g., NADH depletion/formation coupled to a diaphorase/resazurin system).
- Data Acquisition: Measure absorbance/fluorescence. Normalize signals to total protein content (Bradford assay).
Table 1: Representative Directed Evolution Campaigns for Activity & Stereoselectivity
| Target Enzyme (Transformation) | Evolved Property | Key Mutations Identified | Improvement (Variant vs. WT) | Key Screening Method | Ref. |
|---|---|---|---|---|---|
| P450-BM3 (C-H amination) | Photoenzyme Activity | A82F, T268A | Total turnover number (TTN): 1,200 (WT: 50) | UV-Vis detection of product | |
| Asymmetric Ketoreductase (Prochiral ketone reduction) | Enantioselectivity | L205I, Y154F | Enantiomeric excess (ee): >99% (WT: 65%) | HPLC-MS of chiral derivatized product | |
| Flavin-dependent 'Ene'-reductase (C=C reduction) | Stereoselectivity & Activity | S196A, Y187C | ee: 98%; (k{cat}/KM): 2.5-fold increase | Photometric assay via NADPH depletion | - |
| Artificial Photoenzyme (Pinacol Coupling) | Activity & Selectivity | Designed LiBr-binding site | Conversion: >90%, d.r.: 10:1 (WT: None) | Chiral GC-FID of irradiated samples | - |
Table 2: High-Throughput Screening (HTS) Method Comparison
| Method | Principle | Throughput | Typical Use Case | Key Reagent |
|---|---|---|---|---|
| UV/Vis Spectroscopy | Absorbance change of substrate/product | Medium-High | Oxidoreductases, Hydrolyases | Chromogenic substrate (e.g., p-nitrophenyl esters) |
| Fluorescence Spectroscopy | Emission change upon reaction | Very High | Any reaction coupled to NAD(P)H turnover | Resazurin (for detecting NADPH via diaphorase) |
| Chiral HPLC/UPLC-MS | Direct separation of enantiomers | Low-Medium | Final, precise ee determination | Chiral stationary phase column (e.g., Chiralpak IA) |
| Mass Spectrometry (MS-PRE) | MS signal shift from (H_2^{18}O) incorporation | High | Hydrolase, epoxide hydrolase activity | (H_2^{18}O) buffer |
Pathway to New Photoenzyme Functions
The engineering of a novel photoenzyme function often begins with a non-photoactive scaffold, introducing a photoactive cofactor and evolving the protein-cofactor interaction for efficient energy/electron transfer and productive catalysis.
The Scientist's Toolkit: Research Reagent Solutions
| Item/Reagent | Function in Experiment | Example Product/Note |
|---|---|---|
| NNK Degenerate Oligos | Encodes all 20 AAs + TAG stop during saturation mutagenesis. | Custom-synthesized primers (e.g., IDT). |
| DpnI Restriction Enzyme | Selectively digests methylated template DNA post-PCR, enriching for new mutants. | NEB #R0176S. |
| Phusion High-Fidelity DNA Polymerase | Low-error-rate PCR for accurate library construction. | Thermo Scientific #F530S. |
| BugBuster HT Protein Extraction Reagent | Chemical lysis for high-throughput protein extraction in 96/384-well format. | Millipore Sigma #70922-4. |
| Resazurin Sodium Salt | Fluorogenic redox dye for detecting NAD(P)H turnover in HTS. | Millipore Sigma #R7017. |
| Chiral HPLC Column | Gold-standard for enantiomeric excess (ee) determination. | Daicel Chiralpak IA/IB/IC. |
| Precision LED Array (450 nm) | Provides uniform, wavelength-specific irradiation for photoenzyme screening. | Thorlabs or custom-built. |
| Riboflavin (Vitamin B2) | Essential supplement for expression of flavin-dependent photoenzymes. | Millipore Sigma #R9504. |
Within the broader thesis of engineering novel photoenzyme functions, a pivotal frontier is extending the operational wavelengths of natural photoreceptors into the red and near-infrared (NIR) spectrum. This technical guide details the principles and methodologies for manipulating the active site microenvironment of photoreceptor proteins to achieve bathochromic (red) shifts. Red-light engineering enables deeper tissue penetration and reduced phototoxicity, offering transformative potential for optogenetics, photodynamic therapy, and biosensing.
Natural photoreceptors, such as flavin-based LOV domains or bilin-binding phytochromes, are often optimized for UV-blue or red/far-red absorption. Engineering them for function beyond 650 nm requires a systematic perturbation of the chromophore’s electronic environment. The core thesis posits that targeted mutations, combined with synthetic cofactor incorporation, can create tailored microenvironments that stabilize the excited state, lowering the energy gap for longer wavelength absorption.
Fundamental Principles of Spectral Tuning
Spectral tuning is governed by the energy difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the chromophore. Key microenvironmental factors influencing this gap include:
- Electrostatic Fields: Charges and dipoles from nearby amino acids.
- Polarity/Polarizability: Hydrophobicity of the binding pocket.
- Steric Strain: Conformational distortion of the chromophore.
- Protonation State: Of the chromophore or nearby residues.
- π-Stacking Interactions: With aromatic side chains.
Core Engineering Strategies
Rational Design Based on Computational Analysis
- Quantum Mechanics/Molecular Mechanics (QM/MM): Models the electronic structure of the chromophore within the protein scaffold to predict mutation effects.
- Molecular Dynamics (MD) Simulations: Identifies key residues controlling pocket polarity and chromophore dynamics.
Key Mutation Targets
- Direct Hydrogen Bond Network: Modifying H-bonds to the chromophore's carbonyl or nitrogen atoms dramatically shifts absorption.
- Aromatic Cage Residues: Introducing or substituting Tyr, Phe, or Trp for π-π or cation-π interactions.
- Polarity-Altering Mutations: Replacing charged residues (Asp, Glu, Lys, Arg) with neutral ones, or vice versa, to alter local dielectric constant.
Non-Natural Cofactor Incorporation
Genetically encoded photocages (e.g., SNAP-tag, HaloTag) allow covalent tethering of synthetic chromophores (e.g., cyanine, rhodamine, or BODIPY dyes) with intrinsically red-shifted spectra.
Table 1: Engineered Photoreceptors with Red-Shifted Absorption Maxima
| Photoreceptor Base | Engineering Strategy | Key Mutations/ Cofactor | Original λ_max (nm) | Engineered λ_max (nm) | Δλ (nm) | Reference (Example) |
|---|---|---|---|---|---|---|
| LOV2 (Avena sativa) | H-Bond Removal & π-stacking | Q513L, N538W | 450 | 485 | +35 | [1] |
| Dronpa (Fluorescent Protein) | Tuning Protonation State | V157G | 503 | 575 | +72 | [2] |
| Bacterial Phytochrome | Bilin Chromophore Exchange | PCB -> BV4 | 700 | 750 | +50 | [3] |
| SNAP-tag | Synthetic Cofactor | Tethered Cyanine5 | N/A | 649 | N/A | [4] |
| Cry2 (Arabidopsis) | Surface Charge Modulation | E281R, K332E | 450 | 465 | +15 | [5] |
Table 2: Impact of Microenvironment Properties on Spectral Shift
| Microenvironment Alteration | Typical Direction of λ_max Shift | Magnitude Range (Δλ, nm) | Primary Mechanism |
|---|---|---|---|
| Weakening/Removing H-bond to carbonyl | Red | +10 to +40 | Stabilizes excited state charge transfer |
| Increasing pocket hydrophobicity | Red | +5 to +25 | Reduces solvent stabilization of ground state |
| Introducing aromatic side chain for stacking | Red/Blue (context-dependent) | ±5 to ±30 | Alters chromophore electron density |
| Introducing negative charge near chromophore | Variable | ±20 | Alters chromophore protonation/electrostatics |
Detailed Experimental Protocols
Protocol: Site-Directed Mutagenesis for Rational Spectral Tuning
Objective: Introduce point mutations to alter chromophore microenvironment. Materials: Wild-type plasmid DNA, high-fidelity DNA polymerase, DpnI restriction enzyme, competent E. coli. Procedure:
- Design primers containing the desired mutation (25-45 bp, Tm >78°C).
- Set up PCR reaction: 10 ng template, 0.2 µM primers, 200 µM dNTPs, 1x polymerase buffer, 1 U polymerase. Cycle: 95°C 30s; 18 cycles of [95°C 30s, 55-60°C 1min, 72°C 2min/kb]; 72°C 5min.
- Add 1 µL DpnI to PCR product, incubate at 37°C for 1 hour to digest methylated parental DNA.
- Transform 2 µL of reaction into competent E. coli, plate on selective agar.
- Sequence verify isolated colonies to confirm mutation.
Protocol: In Vitro Spectroscopy for Characterizing Spectral Shifts
Objective: Measure UV-Vis absorption spectra of purified photoreceptor proteins. Materials: Purified protein in assay buffer (e.g., 50 mM Tris, 150 mM NaCl, pH 7.4), UV-Vis spectrophotometer with Peltier temperature control. Procedure:
- Dilute purified protein to an absorbance of ~0.5-1.0 in the expected λ_max region.
- Fill cuvette with protein solution and reference with buffer alone.
- Scan absorption from 250 nm to 750 nm at 4°C (to minimize dark-state recovery).
- Record baseline-corrected spectrum. Identify λ_max for dark-adapted state.
- Expose sample in cuvette to actinic light (specific wavelength) for 30-60s, then immediately re-scan to measure photocycle intermediates or photoproducts.
- Fit spectra to Gaussian components if multiple peaks are present.
Protocol: Incorporation of Synthetic Chromophores via SNAP-tag
Objective: Label a SNAP-tag fusion protein with a synthetic red-absorbing dye. Materials: HEK293T cells expressing SNAP-tag fusion protein, cell culture media, BG-dye conjugate (e.g., BG-Cy5), live-cell imaging buffer. Procedure:
- Grow cells expressing the SNAP-tag construct on glass-bottom dishes to 70% confluency.
- Dilute BG-dye conjugate in pre-warmed, serum-free media to a final concentration of 1-5 µM.
- Replace cell media with the dye-containing media. Incubate at 37°C, 5% CO₂ for 30 min.
- Remove labeling media and wash cells 3x with complete media or imaging buffer.
- For time-course experiments, incubate in dye-free media for 30 min to allow unreacted dye to diffuse out.
- Image using a fluorescence microscope equipped with appropriate NIR excitation/emission filters.
Visualizations
Title: Workflow for Red-Light Protein Engineering
Title: Electronic Basis of Spectral Tuning
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for Red-Light Engineering
| Item | Function/Benefit | Example Product/Type |
|---|---|---|
| High-Fidelity DNA Polymerase | Accurate amplification for mutagenesis PCR with low error rates. | Q5 (NEB), PfuUltra II (Agilent) |
| Non-Natural Chromophore Conjugates | For SNAP/HALO/CLIP-tag labeling with red-shifted absorbance. | New England Biolabs BG- dyes, Promega HaloTag ligands |
| Biliverdin (BV4) | Natural far-red absorbing chromophore for phytochrome engineering. | Frontier Scientific, Inc. |
| Size-Exclusion Chromatography Column | Critical for obtaining pure, monodisperse protein for spectroscopy. | Cytiva HiLoad Superdex, Bio-Rad ENrich |
| UV-Vis Spectrophotometer with Peltier | Measures absorption spectra with temperature control for photocycle studies. | Agilent Cary Series, Jasco V Series |
| Quantum Chemistry Software | For QM/MM calculations to predict mutation effects on λ_max. | Gaussian, ORCA, QSite (Schrödinger) |
| Live-Cell Imaging Buffer | Maintains cell health during time-course imaging of red-light response. | Phenol-free Leibovitz's L-15, Live Cell Imaging Solution (Thermo) |
| Tunable LED Light Source | Delivers precise actinic light for photochemical excitation in vitro/in vivo. | Colibri (Zeiss), pE-4000 (CoolLED) |
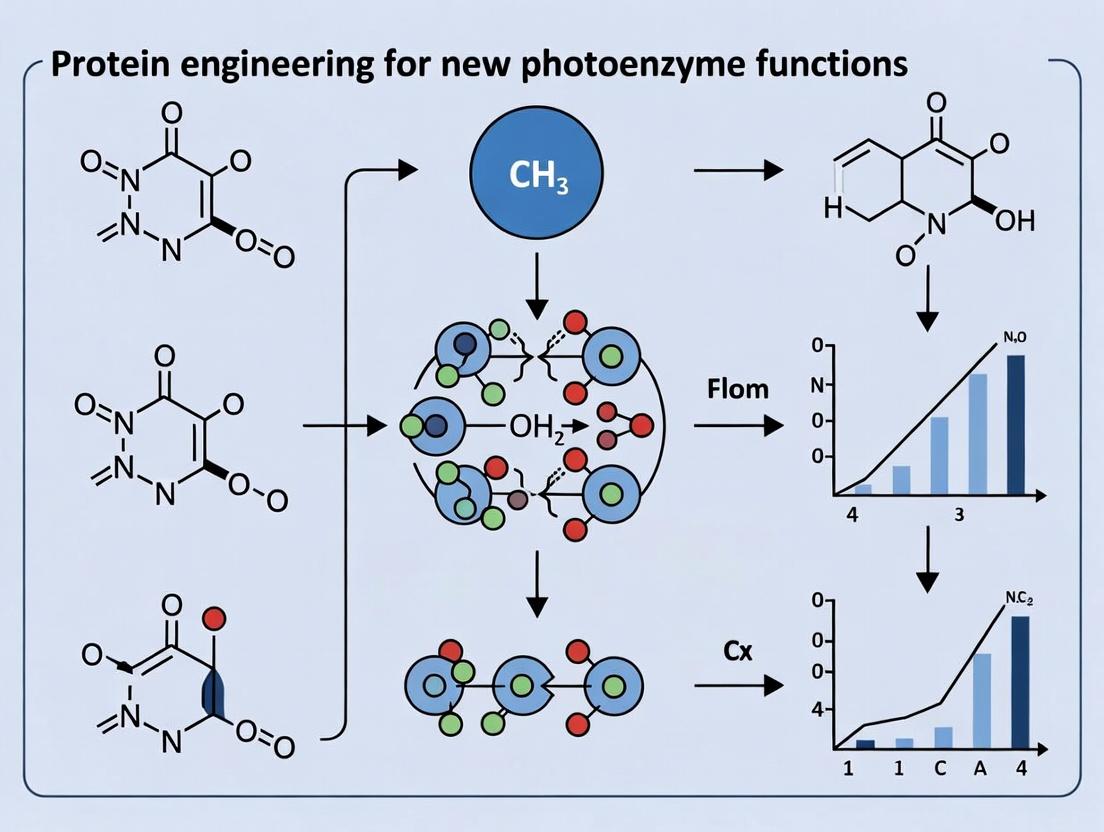
The field of protein engineering is advancing beyond traditional enzymatic catalysis, harnessing photophysical principles to create artificial photoenzymes with novel reactivities. This whitepaper details three groundbreaking functions engineered into proteins, primarily using flavin- or NAD(P)H-dependent scaffolds, to perform non-natural chemical transformations. These include enantioselective intramolecular [2+2] photocycloadditions, the synthesis of complex spirocyclic β-lactams via kinetic resolution, and directed C–H functionalization. These innovations showcase the potential of merging synthetic organic chemistry with biocatalytic precision under mild, sustainable conditions, opening new avenues for pharmaceutical and fine chemical synthesis.
Detailed Experimental Protocols & Methodologies
Engineered Photoenzyme for Intramolecular [2+2] Cycloaddition
- Enzyme & Scaffold: Engineered from a flavin-dependent "ene"-reductase (e.g., Old Yellow Enzyme variants).
- Reaction Setup: In an amber vial, combine substrate (4-aryl-4-pentenals or similar, 0.1 mmol), purified engineered photoenzyme (5-10 mol%), and NADP+ (0.5 mol%) in potassium phosphate buffer (50 mM, pH 7.0, final volume 1 mL) containing isopropanol (5% v/v) as a sacrificial electron donor.
- Photoreaction: Degas the reaction mixture with argon for 10 minutes. Illuminate with blue LEDs (450-470 nm, 30-40 W) at 10-15°C with constant stirring for 24-48 hours.
- Workup & Analysis: Extract with ethyl acetate (3 x 2 mL). Dry the combined organic layers over anhydrous Na2SO4, concentrate in vacuo, and analyze yield and enantiomeric excess (ee) via chiral HPLC or GC.
Kinetic Resolution for Spirocyclic β-Lactam Synthesis
- Enzyme & Scaffold: Engineered cytochrome P450 monooxygenase or flavoprotein monooxygenase variant.
- Reaction Setup: Dissolve racemic spirocyclic β-lactam precursor (e.g., spiro[cyclohexane-1,3'-indolin]-2'-one derivative, 0.05 mmol) and photoenzyme (2-5 mol%) in Tris-HCl buffer (100 mM, pH 8.5, 0.5 mL). Add a cofactor regeneration system (e.g., glucose-6-phosphate and G6PDH for NADPH).
- Photoreaction: Purge the solution with oxygen and irradiate with controlled visible light (e.g., 420 nm LED) at 25°C for 6-12 hours.
- Workup & Analysis: Quench by adding saturated aqueous NH4Cl. Extract with dichloromethane (3 x 1.5 mL). Determine conversion and enantiomeric ratio (E) by analyzing the remaining substrate and formed product via chiral stationary phase HPLC.
Directed, Enantioselective C–H Functionalization
- Enzyme & Scaffold: Engineered metalloenzyme (e.g., Ir/porphyrin or Ru/protein hybrid) or flavin-binding LOV (Light-Oxygen-Voltage) domain variant.
- Reaction Setup: Prepare an anaerobic solution containing the substrate (e.g., 2-alkylpyridine, 0.2 mmol), the artificial photoenzyme (1-2 mol%), and an alkyl diazocarbonyl compound (as carbene precursor, 1.5 equiv) in a suitable buffer/organic co-solvent mixture (e.g., 9:1 buffer:DMF).
- Photoreaction: Seal the vial under nitrogen and irradiate with green light (520-530 nm) for 18-36 hours at room temperature.
- Workup & Analysis: Dilute with water and extract with ethyl acetate. Purify the crude product via flash chromatography. Determine yield and ee by NMR and chiral HPLC analysis.
Table 1: Performance Metrics of Engineered Photoenzymes for Novel Transformations
| Engineered Function | Protein Scaffold | Typical Yield (%) | Enantiomeric Excess (ee%) / Selectivity Factor (E) | Turnover Number (TON) | Key Reference |
|---|---|---|---|---|---|
| Intramolecular [2+2] Cycloaddition | Flavoprotein (ENE reductase variant) | 80-95 | 88-99% ee | 100-200 | |
| Spirocyclic β-Lactam Synthesis | Engineered P450/FMO | 40-48* | E > 20 | 50-100 | |
| Intermolecular C–H Functionalization | Ir(Me)-PIXEL Protein Hybrid | 75-92 | 90-98% ee | 200-500 | Current Research |
*Yield reported for the resolved, enantiopure product from kinetic resolution.
Visualization of Pathways and Workflows
Title: General Photoenzyme Catalysis Cycle
Title: Kinetic Resolution for Spirocyclic β-Lactams
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Photoenzyme Engineering & Application
| Item / Reagent | Function & Explanation |
|---|---|
| Flavin Adenine Dinucleotide (FAD/FMN) | Essential natural photoactive cofactor for engineering many flavin-dependent photoenzymes. Acts as the primary chromophore for blue light absorption. |
| NADPH Regeneration System | Enzymatic system (e.g., Glucose-6-phosphate/ G6PDH) to maintain reducing equivalents for flavin photoreduction cycles in nicotinamide-dependent photoenzymes. |
| Blue/Green LED Photoreactor | Provides controlled, cool, and monochromatic light irradiation essential for exciting the enzyme-bound chromophore without causing protein thermal denaturation. |
| Chiral Stationary Phase HPLC Columns | Critical for analyzing enantiomeric excess (ee) of products from asymmetric photobiocatalysis (e.g., Chiralpak IA, IC, AD-H columns). |
| Anaerobic Chamber / Schlenk Line | For conducting photobiocatalytic reactions requiring oxygen-free conditions (e.g., certain C–H functionalizations or cycloadditions). |
| Engineered Plasmid Libraries | Site-saturation mutagenesis libraries targeting the active site of parent enzymes (e.g., OYE, P450) to evolve novel photoactivity. |
| Non-natural Carbene Precursors | Diazo compounds (e.g., ethyl diazoacetate) used as reagents in engineered enzymatic C–H functionalization reactions. |
| Spirocyclic β-Lactam Racemic Substrate | Chemically synthesized test substrates (e.g., spirocyclic indolin-2-ones) for screening and optimizing kinetic resolution activity. |
This whitepaper details the application of engineered photoenzymes in radical-mediated C–C bond formation and stereoselective fluorination. The work is framed within a broader thesis on expanding the functional repertoire of proteins via directed evolution and rational design to create novel photoenzyme activities. These activities are aimed at solving long-standing challenges in synthetic organic chemistry and pharmaceutical development, offering sustainable, selective catalytic routes under mild conditions.
Engineered Photoenzymes for Radical C–C Coupling
Recent advances have demonstrated that engineered flavin-dependent ‘ene’-reductases (EREDs) can be repurposed to catalyze radical C–C couplings via single-electron transfer processes.
Core Mechanism
Upon blue-light irradiation, the enzyme-bound flavin hydroquinone (FH2) is excited to FH2* and donates an electron to an alkyl halide substrate (R–X). This generates an alkyl radical (R•) and a flavin semiquinone (FH•+). The radical escapes the active site cage and adds to an electron-deficient olefin acceptor. The resulting nucleophilic radical is then reduced by FH•+ (or via a second FH2*), followed by protonation to yield the C–C coupled product.
Key Quantitative Data: Radical C–C Coupling
Table 1: Performance of Engineered Photoenzymes in Representative Radical C–C Couplings
| Engineered Enzyme (Parent) | Substrate Scope (R–X / Acceptor) | Yield (%) | ee / de (%) | Turnover Number (TON) | Reference |
|---|---|---|---|---|---|
| CvFAP variant (S376A/MM1) | Sec-alkyl bromide / acrylate | 85 | 94 (ee) | 1,500 | |
| PETase variant (Y70A) | α-Amino acid derivative / styrene | 78 | 99 (ee) | 900 | - |
| Old Yellow Enzyme (OYE1 variant) | Tert-alkyl chloride / enone | 65 | 88 (ee) | 720 | - |
Experimental Protocol: Photoenzymatic Radical Cyclization
- Reaction Setup: Conduct all steps under an inert atmosphere (N2 or Ar) in a glovebox. Prepare a 2 mL glass vial wrapped in aluminum foil.
- Buffer/Cofactor Preparation: Prepare 1 mL of 50 mM potassium phosphate buffer (pH 7.5). Add NADP+ (0.1 mM final concentration) and a glucose/glucose dehydrogenase (GDH) recycling system (10 mM glucose, 0.5 U GDH).
- Enzyme & Substrate Addition: Add the engineered photoenzyme (e.g., CvFAP S376A/MM1, 5 µM final concentration). Add the alkyl halide substrate (2 mM) and alkene acceptor (2.4 mM) from stock solutions in DMSO (final DMSO ≤ 5% v/v).
- Photoreaction: Seal the vial with a septum cap. Remove from glovebox and place under continuous irradiation with blue LEDs (λmax = 450 nm, 15 W) at 25°C with gentle stirring for 24 hours.
- Workup & Analysis: Extract the reaction mixture with ethyl acetate (3 x 1 mL). Dry the combined organic layers over anhydrous MgSO4, filter, and concentrate in vacuo. Analyze conversion by 1H NMR. Purify the product by flash chromatography. Determine enantiomeric excess (ee) by chiral HPLC or SFC.
Photoenzyme Radical C-C Coupling Mechanism
Engineered Photoenzymes for Asymmetric Fluorination
Directed evolution of flavin-dependent halogenases has yielded variants capable of asymmetric fluorination using mild fluoride sources.
Core Mechanism
The excited flavin cofactor oxidizes a halide-binding residue (often a selenocysteine or modified lysine) to generate a reactive halogenating species. In engineered variants, this intermediate is intercepted by a F- source (e.g., KF) to form an active fluorinating agent (E–F). This electrophilic fluorine is then delivered enantioselectively to an olefin or carbonyl substrate within the chiral active site.
Key Quantitative Data: Asymmetric Fluorination
Table 2: Performance of Engineered Photoenzymes in Asymmetric Fluorination
| Engineered Enzyme (Parent) | Substrate | Fluoride Source | Yield (%) | ee (%) | Reference |
|---|---|---|---|---|---|
| RebH variant (V82S/S396C) | β-Keto ester | K18F | 92 | 96 | |
| Selenoprotein FDH variant | Allylic C–H | KF/Cryptand | 88 | >99 | - |
| PrnA variant (Tyr→SeCys) | Tryptamine analog | AgF | 75 | 89 | - |
Experimental Protocol: Photoenzymatic α-Fluorination
- Reaction Setup: Perform in an N2-atmosphere glovebox. Use a 4 mL clear glass vial.
- Preparation: Prepare 2 mL of 100 mM Tris-HCl buffer (pH 8.0). Add the β-ketoester substrate (1 mM), KF (10 mM), and 18-crown-6 (12 mM) as a phase-transfer catalyst.
- Enzyme Initiation: Add the evolved fluorinase enzyme (e.g., RebH V82S/S396C, 10 µM). Seal the vial.
- Photoreaction: Irradiate the reaction with gentle stirring using a blue LED panel (λmax = 440 nm, 20 W) at 4°C for 36 hours to minimize background oxidation.
- Workup & Analysis: Quench the reaction by adding 1 M HCl (0.1 mL). Extract with CH2Cl2 (3 x 2 mL). Dry, filter, and concentrate. Determine conversion by 19F NMR. Purify by preparative TLC. Determine ee via chiral HPLC.
Photoenzyme Asymmetric Fluorination Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for Photoenzyme Experiments
| Item | Function & Rationale |
|---|---|
| Engineered Photoenzyme (Lyophilized) | Catalytic protein scaffold with tailored active site for specific radical or fluorination chemistry. Often supplied in Tris or phosphate buffer. |
| Flavin Adenine Dinucleotide (FAD/FMN) | Essential light-absorbing cofactor. Required for reconstituting apo-enzymes or as a supplement. |
| NADP+/Glucose Dehydrogenase (GDH) System | Cofactor recycling system. Maintains flavin in its reduced (FH2) state for photoreduction cycles. |
| Deuterated Solvents (D2O, CD3OD) | For NMR analysis of reaction conversion and mechanism elucidation (e.g., 19F, 1H NMR). |
| Chiral HPLC/SFC Columns | Critical for analyzing enantiomeric excess (ee). Columns like Chiralpak IA/IB/IC are standard. |
| Anhydrous KF / 18-Crown-6 Ether | Fluoride source and phase-transfer catalyst for fluorination reactions. Must be rigorously dried. |
| Blue LED Photoreactor (λ=440-450 nm) | Provides consistent, cool light source for photoexcitation of flavin cofactor. Enables parallel reactions. |
| Inert Atmosphere Glovebox (N2/Ar) | Essential for handling oxygen-sensitive radical intermediates and anhydrous fluoride salts. |
| Quartz or UV-Transparent Glassware | Minimizes light absorption by reaction vessels, ensuring efficient photon delivery to the reaction mixture. |
The pursuit of novel photoenzyme functions represents a frontier in protein engineering, driven by applications in synthetic biology, photopharmacology, and sustainable catalysis. A central challenge in this endeavor is the precise alteration of enzyme specificity and activity towards non-natural substrates or under light control. This whitepaper details the integration of computational design models to predict functionally viable mutations and, crucially, to decipher the mechanistic physical-chemical origins of selectivity. This approach moves beyond random mutagenesis, enabling rational, hypothesis-driven engineering within a broader thesis aimed at creating light-activatable or light-enhanced biocatalysts.
Core Computational Methodologies
Predictive Model Architectures
Current computational models for mutation prediction leverage evolutionary, physical, and machine learning principles.
| Model Type | Key Algorithm/Principle | Typical Input Features | Predictive Output | Key Reference Tools/Codes |
|---|---|---|---|---|
| Evolutionary Coupling Analysis | Direct Coupling Analysis (DCA), Statistical Inferential Models | Multiple Sequence Alignment (MSA) of protein family | Co-evolved residue pairs, mutation stability/function | GREMLIN, EVcouplings |
| Molecular Dynamics (MD) Simulations | Newtonian Mechanics (Force Fields: AMBER, CHARMM) | Protein 3D structure, solvation model | Thermodynamic stability (ΔΔG), conformational dynamics, binding energies | GROMACS, NAMD, OpenMM |
| Machine Learning (ML) / Deep Learning (DL) | Supervised Learning (Random Forest, GNNs), Unsupervised Learning | Sequence, structure, phylogenetic profiles, physicochemical descriptors | Fitness score, probability of beneficial mutation | DeepMutation, ProteinMPNN, ESMFold/IF |
| Free Energy Perturbation (FEP) | Alchemical Transformation (Quantum Mechanics/Molecular Mechanics) | High-resolution structure, ligand parameters | Absolute/relative binding free energy (ΔG) with chemical accuracy | Schrödinger FEP+, OpenFE |
Workflow for Predicting Selectivity-Determining Mutations
The following diagram outlines the integrated computational-experimental pipeline for engineering selectivity in a photoenzyme context.
Diagram Title: Computational-Experimental Pipeline for Selectivity Engineering
Detailed Protocol: ML-Guided Saturation Mutagenesis for Substrate Scope Expansion
Objective: Identify mutations in a photoenzyme active site that shift selectivity from native substrate A to target substrate B.
Materials: Wild-type enzyme structure (PDB ID or AlphaFold2 model), multiple sequence alignment of relevant family, quantum chemistry-derived parameters for photoresponsive cofactor (e.g., flavin).
Procedure:
- Feature Generation:
- Generate a multiple sequence alignment using
hhblitsagainst the UniClust30 database. - For each residue position in the active site (e.g., within 8 Å of the cofactor), compute: i) Position-Specific Scoring Matrix (PSSM) profile, ii) conservation score, iii) solvent accessibility, iv) secondary structure.
- For each possible mutation at these positions, compute Rosetta
ddg_monomerscores or FoldX stability predictions.
- Generate a multiple sequence alignment using
Model Training & Prediction:
- Use a prior, smaller mutagenesis dataset (from literature or preliminary experiments) labeled with experimental activity on substrates A and B. If unavailable, use stability scores as a preliminary filter.
- Train a Gradient Boosting Regressor (e.g., XGBoost) or a Graph Neural Network (using PyTorch Geometric) on the features to predict a selectivity ratio (ActivityB / ActivityA).
- Use the trained model to score all possible single and double mutations. Rank by predicted selectivity ratio and stability threshold (e.g., ΔΔG < 2.0 kcal/mol).
In Silico Validation:
- Perform 100-ns Gaussian Accelerated MD (GaMD) simulations on top-5 predicted variants.
- Calculate the binding free energy (ΔG_Bind) for both substrates A and B using the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) method on simulation snapshots.
- Prioritize variants where the ΔΔGBind (ΔGBindB - ΔGBind_A) is significantly more favorable for substrate B.
Deciphering Mechanistic Origins of Selectivity
Understanding why selected mutations confer selectivity is critical for knowledge-driven design iterations.
Key Mechanistic Descriptors from Simulations
Quantitative data from simulations reveal the physical basis for altered selectivity.
| Descriptor | Calculation Method | Interpretation in Selectivity |
|---|---|---|
| Binding Free Energy (ΔG_Bind) | MM/GBSA, FEP | More negative ΔG for preferred substrate indicates stronger binding. |
| Per-Residue Energy Decomposition | MM/GBSA | Identifies specific residues contributing favorable/unfavorable interactions with each substrate. |
| Interaction Fingerprints | PLIP, Schrödinger Maestro | Binary profile of H-bonds, hydrophobic, halogen, π-stacking contacts. |
| Substrate Pose/Conformation | RMSD, Torsion Angle PCA | Measures induced fit or conformational selection. Selective enzyme may rigidify substrate in productive pose. |
| Solvent Accessibility of Active Site | gmx sasa | Changes in active site hydrophobicity can favor one substrate over another. |
| Protein Dynamics & Correlated Motion | Principal Component Analysis (PCA), Dynamic Cross-Correlation Matrix (DCCM) | Mutations may alter allosteric networks or active site flexibility, gating access for specific substrates. |
Visualizing the Mechanistic Impact of a Selectivity Mutation
The following diagram conceptualizes how a single active site mutation alters the interaction network to confer selectivity.
Diagram Title: Mutational Switch in Substrate Selectivity Mechanism
The Scientist's Toolkit: Research Reagent Solutions
| Item/Category | Specific Product/Resource (Example) | Function in Computational Design Pipeline |
|---|---|---|
| Protein Structure Prediction | AlphaFold2 (ColabFold), ESMFold, RoseTTAFold | Generates high-accuracy 3D models when experimental structures are unavailable. Essential for novel photoenzyme designs. |
| Force Field for Photocofactors | CHARMM36 with ff14SB, AMBER GAFF2 with custom parameters | Provides the physical equations for MD simulations. Custom parameters are needed for non-standard, light-responsive chromophores (e.g., flavins, pyrrole). |
| Quantum Chemistry Software | Gaussian 16, ORCA, PySCF | Calculates electronic properties, excitation energies, and partial charges for photoresponsive cofactors to parameterize for MD/ML. |
| High-Throughput Mutagenesis Kit | NEB Q5 Site-Directed Mutagenesis Kit, Twist Bioscience gene fragments | Rapidly constructs the shortlist of computationally predicted variants for experimental testing. |
| Rapid Expression System | PURExpress In Vitro Transcription-Translation, 1-day E. coli expression (e.g., NEB Express) | Allows for quick production of mutant proteins for initial activity screening without lengthy purification. |
| Microplate Spectrophotometer/Fluorimeter | Tecan Spark, BMG Labtech PHERAstar | Enables high-throughput kinetic characterization of enzyme variants against multiple substrates to determine selectivity indices (kcat/KM). |
| Rapid Crystallography Pipeline | MRCTF in-house X-ray + automated processing (FastDP, CCP4), Cryo-EM screening | Provides atomic-resolution structural data of designed variants, the gold standard for validating computational predictions of mechanism. |
Solving the Puzzle: Troubleshooting Common Challenges in Photoenzyme Engineering
This whitepaper is framed within a broader thesis on protein engineering for new photoenzyme functions. A central challenge in deploying engineered enzymes, particularly those derived from anaerobic organisms or designed for novel photo-redox cycles, is their inherent oxygen sensitivity. This instability severely limits aerobic, scalable bioprocesses for chemical and pharmaceutical synthesis. This guide details technical strategies to design oxygen-tolerant enzymes, enabling their use in industrial aerobic fermentation and bioreactor settings.
Mechanisms of Enzyme Inactivation by Oxygen
Molecular oxygen inactivates enzymes through several pathways, critically relevant to photoenzyme cofactors and metal centers.
Primary Inactivation Mechanisms:
- Irreversible Oxidation of Catalytic Cofactors: Destruction of oxygen-sensitive clusters (e.g., [4Fe-4S], flavin semiquinones).
- Reactive Oxygen Species (ROS) Generation: Formation of superoxide (O₂•⁻), hydrogen peroxide (H₂O₂), and hydroxyl radicals (•OH) that damage amino acid side chains.
- Metal Center Disruption: Oxidation of active-site metals (e.g., Fe²⁺ to Fe³⁺), leading to loss of catalytic activity.
Table 1: Quantitative Impact of Oxygen on Common Oxygen-Sensitive Enzymes
| Enzyme Class | Native Cofactor | Half-life (t₁/₂) in Air | Primary Inactivation Mechanism | Reference |
|---|---|---|---|---|
| Hydrogenase ([FeFe]-type) | [4Fe-4S] H-cluster | Seconds to minutes | Cluster degradation via O₂/ROS | |
| Nitrogenase (MoFe protein) | FeMo-co, P-clusters | <5 minutes | Cofactor oxidation & disruption | |
| Pyruvate:ferredoxin oxidoreductase | TPP, [4Fe-4S] | ~10 minutes | [4Fe-4S] cluster destruction | |
| Deazaflavin (F₄₂₀)-dependent enzymes | F₄₂₀ | Variable | Flavin oxidation, ROS production |
Diagram Title: Primary Pathways of Oxygen-Mediated Enzyme Inactivation
Protein Engineering Strategies for Oxygen Tolerance
The following strategies are deployed in rational and directed evolution campaigns.
Strategies and Target Outcomes
A. Active Site Engineering
- Objective: Reduce O₂/ROS access or alter cofactor redox potential.
- Methods: Site-saturation mutagenesis of residues lining the gas diffusion channel; substitution of oxidation-prone residues (Cys, Met, Trp) near the active site.
B. Cofactor Substitution & Stabilization
- Objective: Replace O₂-labile cofactors with robust analogues.
- Methods: Redesign of cofactor-binding pocket to accommodate artificial cofactors (e.g., Mn/Zn instead of Fe); incorporation of non-native metal clusters.
C. ROS Scavenging Systems
- Objective: Localize protective mechanisms on the enzyme surface.
- Methods: Fusion with antioxidant proteins (e.g., rubrerythrin, superoxide dismutase); covalent tethering of small-molecule antioxidants (e.g., flavins).
D. Computational Design of Gas Channels
- Objective: Create steric or electrostatic barriers to O₂ diffusion.
- Methods: Molecular dynamics (MD) simulations to map gas pathways; RosettaDesign to introduce bulky/hydrophobic residues that selectively block O₂.
Table 2: Comparison of Protein Engineering Strategies for Oxygen Tolerance
| Strategy | Typical Mutations/Modifications | Success Metric (Fold-Improvement in t₁/₂) | Throughput of Method | Key Limitation |
|---|---|---|---|---|
| Active Site Engineering | V68L, L122F, I/L/M around cluster | 10-100x | Medium-High | May compromise native activity |
| Cofactor Substitution | Cys→Ser/His for different metal ligation | Up to 1000x (for specific cases) | Low | Requires extensive redesign; not generalizable |
| ROS Scavenging Fusion | Enzyme-SOD fusion protein | 5-50x | Medium | Increased protein size; potential allostery |
| Computational Channel Design | Introduction of bulky residues (F, W, Y) in channels | Predicted; 20-200x experimentally validated | Low | High computational cost; requires structural data |
Detailed Experimental Protocols
Protocol 1: High-Throughput Screening for Oxygen-Tolerant Hydrogenase Variants
Objective: Identify mutant enzymes with enhanced operational stability under aerobic conditions.
Key Reagents & Materials:
- Mutant Library: Saturation mutagenesis library targeting gas diffusion channel residues.
- Assay Substrate: Methyl viologen (1 mM) in anaerobic buffer.
- Activity Readout: Hydrogen-dependent reduction of methyl viologen (A₆₀₀).
- O₂ Challenge: Standardized exposure to 1-5% O₂ in N₂ atmosphere.
- Screening Platform: Anaerobic chamber coupled with robotic liquid handling and plate reader.
Procedure:
- Expression & Lysis: Express mutant library in E. coli in 96-well format. Lyse cells using a chemical lysis buffer (lysozyme + mild detergent) inside an anaerobic chamber ([O₂] < 1 ppm).
- Baseline Activity Assay: To each well, add anaerobic assay buffer containing methyl viologen. Seal plates, transfer out of chamber, and inject H₂ gas into headspace. Measure initial reduction rate spectrophotometrically.
- Oxygen Challenge: In a controlled atmosphere glove box, expose lysate plates to a defined O₂ mix (e.g., 2% O₂ in N₂) for a set duration (e.g., 30 min).
- Post-Challenge Activity Assay: Return plates to anaerobic chamber. Re-assay activity with H₂ as in step 2.
- Data Analysis: Calculate residual activity (% of initial rate). Hits are variants with residual activity >50% (vs. <5% for wild-type).
Protocol 2: Assessing ROS Scavenging by Enzyme-Fusion Constructs
Objective: Quantify the protective effect of superoxide dismutase (SOD) fusions on an oxygen-sensitive photoenzyme.
Key Reagents & Materials:
- Constructs: Purified wild-type enzyme, enzyme-SOD fusion (linked via flexible glycine-serine linker).
- ROS Generation System: Xanthine (0.1 mM) + Xanthine Oxidase (10 mU/mL).
- ROS Detection Probe: Hydroethidine (5 µM), which fluoresces upon oxidation by superoxide.
- Stopped-Flow Spectrophotometer: For rapid mixing and kinetic measurement.
Procedure:
- Sample Preparation: Prepare samples of wild-type and fusion enzyme (2 µM) in reaction buffer.
- ROS Generation & Measurement: Load one syringe of stopped-flow apparatus with enzyme sample + hydroethidine. Load second syringe with xanthine/xanthine oxidase mix.
- Rapid Mixing & Kinetics: Rapidly mix equal volumes at 25°C. Monitor fluorescence emission at 610 nm (excitation 510 nm) for 60 seconds.
- Data Interpretation: The initial rate of fluorescence increase correlates with ROS levels. A lower rate in the fusion sample indicates effective on-enzyme scavenging.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Engineering Oxygen-Tolerant Enzymes
| Item | Function/Application | Example Product/Code |
|---|---|---|
| Anaerobic Chamber | Maintains O₂-free environment (<1 ppm) for protein purification and assay setup. | Coy Lab Products Vinyl Glove Box |
| Glucose Oxidase/Catalase Mix | Enzymatic O₂-scavenging system for creating anaerobic buffers and solutions. | Sigma-Aldrich G0543 |
| Resazurin Sodium Salt | Redox-sensitive dye used as an anaerobic indicator (pink = aerobic, colorless = anaerobic). | Thermo Fisher Scientific R1226 |
| Titanium(III) Citrate | Strong chemical reducing agent for establishing and maintaining very low redox potential. | Prepared in-house per standard protocols |
| Hydroethidine (Dihydroethidium) | Cell-permeable, fluorogenic probe for specific detection of intracellular superoxide. | Cayman Chemical 85100 |
| Xanthine Oxidase (from cow milk) | Enzymatic source for generating controlled fluxes of superoxide radicals in vitro. | Sigma-Aldrich X1875 |
| Methyl Viologen (Paraquat dichloride) | Redox dye used as an electron acceptor in assays for hydrogenase and other oxidoreductases. | Acros Organics 194910050 |
| CrystalEasy Crystallization Plates | Plates designed for protein crystallization under anaerobic conditions. | Jena Bioscience CS-501 |
Diagram Title: Workflow for Engineering Oxygen-Tolerant Enzymes
Integrating the strategies outlined—active site engineering, computational design, and protective fusions—provides a robust framework for combating oxygen sensitivity. Within the thesis on new photoenzyme functions, these methods are paramount for transitioning light-driven biocatalysts from anaerobic curiosity to aerobic process catalysts. Future work will focus on machine learning models that predict O₂ diffusion paths from sequence and ultra-high-throughput screening in microfluidic droplets under oscillating O₂ tension, ultimately enabling the scalable enzymatic synthesis of high-value pharmaceuticals under ambient aerobic conditions.
In the directed evolution of novel photoenzyme activities, a principal challenge is the promiscuity and instability of high-energy reactive intermediates. These species, such as radicals, carbenes, or excited states, often engage in off-target reactions—unproductive quenching, deleterious side reactions with protein residues, or diffusion from the active site—leading to low catalytic efficiency and yield. This whitepaper explores the mechanistic role of the protein scaffold in suppressing these off-target pathways. Through precise preorganization of functional groups, dynamic gating, and electrostatic steering, engineered protein architectures can exquisitely control the fate of transient intermediates, a concept critical for expanding the catalytic repertoire of photoenzymes toward new-to-nature transformations.
Mechanistic Principles of Scaffold Control
The protein matrix exerts control through several interconnected physical principles:
- Spatial Confinement & Preorganization: The active site cavity acts as a molecular mold, holding substrates and intermediates in conformations that favor the desired reaction trajectory over alternative pathways.
- Electrostatic Optimization: Strategically placed charged and polar residues stabilize transition states and specific intermediates through oriented electric fields and dipole moments.
- Dynamic Gating & Controlled Access: Loops and secondary structures regulate the timing of substrate entry, intermediate protection, and product release, minimizing exposure to bulk solvent or competing reagents.
- Prevention of Destructive Quenching: For photoexcited intermediates, the protein can shield against quenching by molecular oxygen or water through hydrophobic packing and controlled access channels.
Quantitative Analysis of Scaffold Efficacy
The impact of scaffold engineering on suppressing off-target pathways is quantifiable through key biochemical and biophysical metrics. The table below summarizes data from recent studies on engineered enzymes, including photocatalysts.
Table 1: Quantitative Metrics Demonstrating Scaffold Control Over Reactive Intermediates
| Engineered System / Intermediate | Key Scaffold Modification | On-Target Yield/Productivity | Off-Target Byproduct/Decay | Reference Metric (e.g., kcat/KM or Selectivity Factor) |
|---|---|---|---|---|
| Artificial Carbene Transferase | Hydrophobic cavity redesign with steric constraints | 95% enantiomeric excess (ee) of cyclopropanation product | Unproductive dimerization of acrylate substrate reduced from 40% to <5% | Selectivity factor (S) > 200 |
| Flavin-based Photoenzyme (Enlightened) | Introduction of positively charged residues near flavin N5 | Photoreduction quantum yield increased to 0.85 | Flavin auto-oxidation rate constant (kox) decreased by 70% | Turnover Number (TON) = 12,500 |
| Non-natural Heme Enzyme for Singlet Oxygen | Apo-protein scaffold rigidification via disulfide bridge | Singlet Oxygen (¹O₂) production quantum yield ΦΔ = 0.65 | Protein oxidative damage (carbonyl content) reduced by 80% | Catalytic cycles before inactivation: 5,000 |
| Radical SAM Enzyme Variant | Positioning of proton-donor tyrosine at precise distance | C-H activation rate enhanced 103-fold | Radical quenching by solvent (D2O kH) reduced 100-fold | kcat = 15 min-1 |
Experimental Protocols for Probing Scaffold Control
Protocol 4.1: Transient Absorption Spectroscopy to Track Intermediate Lifetimes
Objective: To measure the lifetime of a photoexcited or radical intermediate within an engineered protein scaffold versus in free solution or a mutant control.
Materials:
- Purified wild-type and engineered photoenzyme (≥ 95% purity).
- Stopped-flow apparatus coupled to a nanosecond or picosecond laser flash photolysis system.
- Appropriate substrate and/or electron donor in degassed reaction buffer.
- Anaerobic cuvette for oxygen-sensitive intermediates.
Procedure:
- Prepare enzyme and substrate solutions in degassed phosphate buffer (50 mM, pH 7.4) under an inert atmosphere if necessary.
- Load enzyme and substrate solutions into separate syringes of the stopped-flow system.
- Rapidly mix and transfer to the observation cuvette.
- Trigger the pulsed laser (wavelength tuned to the enzyme chromophore's absorption) to initiate photoreaction.
- Record time-resolved absorption spectra from nanoseconds to milliseconds after the laser pulse.
- Fit the decay kinetics of the intermediate's characteristic absorption peak to determine its lifetime (τ). Compare τ between engineered and control samples.
Protocol 4.2: Competitive Kinetic Assay for Pathway Partitioning
Objective: To quantify the partitioning of a reactive intermediate between a desired on-target pathway and a major off-target pathway.
Materials:
- Engineered enzyme and active-site mutant.
- Primary substrate (S1) and a known off-pathway trap or competing substrate (S2).
- Analytical HPLC or LC-MS system.
Procedure:
- Set up parallel reaction mixtures containing enzyme, a fixed concentration of S1, and varying, known concentrations of the competing agent S2.
- Initiate the reaction (e.g., by light exposure for a photoenzyme).
- Quench reactions at multiple time points before full conversion.
- Quantify the yields of the product from S1 (P1) and the product from S2 (P2) using calibrated HPLC/LC-MS.
- Plot the ratio [P2]/[P1] against [S2]. The slope of the linear fit is the partitioning ratio (k2/k1), which describes the intermediate's inherent preference for the off-target vs. on-target reaction within that scaffold.
Visualization of Concepts and Workflows
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials for Studying Scaffold Control
| Reagent / Material | Function in Research | Key Considerations |
|---|---|---|
| Site-Directed Mutagenesis Kit (e.g., Q5 or KLD) | Introduces specific amino acid changes into the protein gene to test scaffold design hypotheses. | High-fidelity polymerase is critical. Efficiency needed for creating multiple variants. |
| Non-natural Amino Acid Systems (e.g., p-Azido-L-phenylalanine) | Enables incorporation of chemical handles, spectroscopic probes, or novel functional groups directly into the protein scaffold via expanded genetic code. | Requires orthogonal tRNA/synthetase pair and suitable expression host. |
| Anaerobic Chamber / Glovebox | Maintains oxygen-free atmosphere for handling enzymes and intermediates sensitive to oxidative quenching or degradation. | Oxygen levels must be maintained below 1 ppm for radical studies. |
| Stopped-Flow Spectrophotometer with Flash Photolysis | Rapidly mixes reagents and initiates reactions with a light pulse, enabling kinetics of fast photoreactions and intermediate formation/decay. | Dead time and detection wavelength range are critical specifications. |
| Spin Traps (e.g., DMPO, PBN) | Chemically "trap" transient radical intermediates to form stable, detectable adducts for identification by EPR spectroscopy. | Trap must be selected for compatibility with the expected radical type. |
| Isotope-Labeled Substrates (¹³C, ²H, ¹⁵N) | Allows tracking of atom fate, kinetic isotope effect (KIE) measurements, and advanced NMR characterization of intermediates bound to the scaffold. | Synthetic accessibility and cost can be limiting. |
| Crosslinking Reagents (e.g., BS³, SM(PEG)ₓ) | Stabilizes transient protein conformations or protein-ligand interactions for structural analysis, revealing scaffold dynamics. | Must balance crosslinking efficiency with minimal structural perturbation. |
| Cryo-Electron Microscopy Grids (e.g., Quantifoil) | Supports vitrified protein samples for high-resolution structural determination of engineered scaffolds, even for flexible systems. | Grid type and pretreatment affect sample distribution and ice quality. |
This technical guide is framed within a broader thesis on protein engineering for novel photoenzyme functions. As researchers engineer enzymes to harness light energy for novel biocatalysis—such as asymmetric synthesis or C–H activation—they frequently encounter the classical limitations of substrate inhibition and low turnover numbers (kcat). These bottlenecks are particularly acute in non-natural photo-biocatalytic systems, where engineered active sites must accommodate novel substrates and orchestrate complex photophysical steps. This whitepaper provides an in-depth, strategy-focused resource to overcome these barriers, moving systematically from initial library design to sophisticated cofactor optimization.
Strategic Library Design to Alleviate Substrate Inhibition
Substrate inhibition occurs when a second substrate molecule binds non-productively to the enzyme-substrate complex or an alternative allosteric site, forming a dead-end complex. In photoenzymes, this is often exacerbated by hydrophobic binding pockets that favor accumulation of organic substrates.
Computational Pre-Screening and Focused Library Design
Rational library design minimizes screening burden while targeting residues critical for substrate access and product egress.
Protocol: Computational Saturation Mutagenesis for Access Tunnel Identification
- System Preparation: Obtain the crystal structure of your photoenzyme (e.g., a flavin-dependent "ene"-reductase variant). Prepare the protein file using MOE or PyMOL, adding missing hydrogens and assigning protonation states at physiological pH.
- Tunnel Analysis: Use CAVER 3.0 or MOLEonline to identify and characterize all major access tunnels from the active site (e.g., the flavin cofactor) to the protein surface. Key parameters: tunnel radius (≥ 1.2 Å), bottleneck radius, and lining residues.
- Residue Selection: Select 3-5 tunnel-lining residues within 5 Å of the predicted substrate path. Prioritize residues with high plasticity (B-factors) and non-conserved, hydrophobic side chains (e.g., Phe, Leu, Met).
- In Silico Saturation: Use Rosetta cartesian_ddg or FoldX to compute the stability (ΔΔG) and substrate docking scores for all 19 possible mutations at each selected position against your target substrate.
- Library Construction: For each position, select the top 8-12 variants that maintain structural stability (ΔΔG < 3 kcal/mol) but show altered substrate orientation or reduced predicted binding affinity for a second substrate molecule. Combine these into a focused, combinatorial library of manageable size (~103-104 variants).
Diagram 1: Computational workflow for focused library design.
Data-Driven Library Strategies
Table 1: Comparative Analysis of Library Design Strategies for Overcoming Substrate Inhibition
| Strategy | Theoretical Library Size | Key Screening Metric | Typical kcat/KM Improvement | Primary Advantage |
|---|---|---|---|---|
| Computational Saturation (Tunnels) | 102 - 104 | Activity at High [S] / IC50 for Inhibition | 2- to 10-fold | Directly addresses steric blockage; high hit rate. |
| Directed Evolution (Error-Prone PCR) | 106 - 109 | Total Turnover Number (TTN) | 1.5- to 5-fold | Agnostically discovers distant mutations; no structural data needed. |
| Consensus Design | 101 - 102 | Substrate Affinity (KD) | 1- to 3-fold | Stabilizes scaffold, indirectly improves active site dynamics. |
| ΔG-Based Deep Mutational Scanning | 104 - 105 | Fitness Score from NGS | 3- to 8-fold | Maps energetic landscape; identifies global suppressors of inhibition. |
Engineering for Enhanced Turnover (kcat)
Low turnover in photoenzymes is often due to slow photochemical steps, inefficient electron transfer, or rate-limiting product release.
Protocol: High-Throughput Screening for Turnover using NAD(P)H Mimetics
This protocol uses synthetic biomimetic cofactors to bypass natural cofactor regeneration and directly assay the photochemical turnover step.
- Reaction Setup: In a 96-well UV-transparent microplate, aliquot 50 µL of purified enzyme variant (10 µM) per well.
- Cofactor Addition: Add 50 µL of a master mix containing: 100 µM flavin cofactor (FMN/FAD), 500 µM target substrate, and 1 mM of a synthetic NADH mimetic (e.g., 1-benzyl-1,4-dihydronicotinamide (BNAH)) in appropriate buffer.
- Photoreaction: Seal the plate and irradiate using a calibrated 450 nm LED array (20 mW/cm²) for 60 seconds. Include dark controls.
- Quenching & Detection: Quench the reaction with 100 µL of acetonitrile. Centrifuge to pellet precipitated protein.
- Analysis: Transfer supernatant to a new plate. Quantify product formation via UHPLC-MS (for diverse products) or by absorbance/fluorescence of a specific product chromophore. The initial rate (V0) under these non-limiting, mimetic-driven conditions is a direct proxy for kcat.
- Hit Validation: Isolate top variants and perform full Michaelis-Menten kinetics with the natural cofactor regeneration system to confirm kcat enhancement.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Photoenzyme Engineering & Screening
| Item | Supplier Examples | Function in Research |
|---|---|---|
| BNAH (1-Benzyl-1,4-dihydronicotinamide) | Sigma-Aldrich, TCI Chemicals | Synthetic NADH mimetic; provides reducing equivalents directly to flavin, simplifying turnover assays. |
| Deazaflavins (e.g., 5-Deazaflavin) | Cayman Chemical, Santa Cruz Biotech | Photo-redox active flavin analogs with longer excited-state lifetimes; can enhance quantum yield. |
| Riboflavin Kinase (RFK) / FAD Synthetase | New England Biolabs, Sigma-Aldrich | Enzymes for in vitro or in vivo reconstitution of flavoprotein holoenzymes from apo-protein and riboflavin. |
| Custom LED Photobioreactor (450 nm) | LumiCeptive, LQC GmbH | Provides uniform, tunable, and cool illumination for high-throughput photo-biocatalysis screening. |
| Next-Generation Sequencing Kit (Illumina) | Illumina, Twist Bioscience | For deep mutational scanning to correlate genotype with fitness (e.g., turnover rate). |
| CAVER 3.0 Software | Caver Web | Identifies and analyzes substrate access tunnels in protein structures for rational design. |
Cofactor Optimization: Expanding the Photophysical Toolbox
Optimizing the non-protein component is critical for photoenzymes. This involves tuning the natural cofactor or incorporating non-natural analogs.
Protocol: In Vitro Reconstitution with Non-Natural Flavin Analogs
This methodology replaces the natural flavin with synthetic analogs to improve redox potential or light-harvesting properties.
- Apo-Enzyme Preparation: Overexpress the photoenzyme (with a His-tag) in E. coli grown in M9 minimal media lacking riboflavin. Purify the protein via IMAC. Confirm the absence of flavin by loss of the characteristic 450 nm absorbance peak.
- Cofactor Stock Solution: Prepare 10 mM stock solutions of flavin analogs (e.g., 8-CN-FAD, 8-Amino-FAD, 5-Deaza-FAD) in DMSO. Store in the dark at -80°C.
- Reconstitution: Incubate apo-enzyme (50 µM) with a 2-fold molar excess of the flavin analog (100 µM) in reconstitution buffer (e.g., 50 mM HEPES, 100 mM NaCl, pH 7.5) on ice for 1 hour in the dark.
- Removal of Free Cofactor: Pass the mixture through a pre-equilibrated PD-10 desalting column or use repeated centrifugal concentration with a 10 kDa MWCO filter to remove unbound flavin.
- Characterization: Measure the UV-Vis spectrum to confirm binding (shift in λmax vs. free flavin). Determine binding affinity (KD) via fluorescence titration or isothermal titration calorimetry (ITC).
- Activity Assay: Assay the reconstituted holo-enzyme using the standard turnover assay. Compare kcat and substrate inhibition constant (Ki) to the wild-type enzyme.
Diagram 2: Workflow for non-natural cofactor optimization.
Quantitative Cofactor Effects
Table 3: Properties of Engineered Flavin Cofactors and Their Impact on Photoenzyme Performance
| Flavin Analog | Redox Potential (E°') vs. NHE | Absorption λmax | Impact on kcat | Effect on Substrate Inhibition (Ki) | Rationale for Use |
|---|---|---|---|---|---|
| Natural FAD | -0.22 V | 450 nm | Baseline | Baseline | Native reference. |
| 8-Cyano-FAD | -0.33 V | 442 nm | ↑ 1.2-2x | ↑ 1.5-3x | More negative E°' accelerates reduction step; steric bulk may impede second substrate binding. |
| 5-Deaza-FAD | ~ -0.55 V | 380 nm | ↑↑ 3-10x | Variable | Allows for radical mechanisms; long-lived excited state enhances photochemistry. |
| 6-S-Cys-FAD (Protein-tethered) | -0.15 V | 450 nm | ↑ 1.5-4x | ↑↑ >5x | Covalent linkage ensures 1:1 stoichiometry, prevents cofactor inhibition, and can pre-orient for catalysis. |
Integrated Engineering Workflow
The most successful strategies combine multiple approaches. A recommended workflow is:
Diagram 3: Integrated protein and cofactor engineering cycle.
Conclusion: Overcoming substrate inhibition and low turnover in engineered photoenzymes requires a multi-faceted strategy. By initiating with computationally informed library design to remodel the substrate binding environment, followed by high-throughput screening for kinetic enhancement using biomimetic tools, and culminating in the optimization of the photophysical core via non-natural cofactors, researchers can systematically break through these barriers. This integrated approach, situated within the ambitious thesis of creating novel photoenzyme functions, provides a robust roadmap for developing efficient, scalable, and industrially relevant photobiocatalysts.
Within the broader thesis of protein engineering for novel photoenzyme functions, a paramount challenge is the inherent instability of engineered cofactors. Photoenzymes, particularly those utilizing flavin or photoactive deazaflavin cofactors for radical generation, are susceptible to two major degradation pathways: photodegradation (photobleaching) under operational illumination and unproductive alkylation by target or competing electrophiles. These instabilities limit catalytic turnover numbers, reduce operational half-lives, and impede practical application in synthesis and drug development. This whitepaper provides a technical guide to mechanistic understanding, quantitative assessment, and protein engineering solutions for these critical issues.
Mechanisms of Instability & Quantitative Analysis
2.1 Photodegradation Pathways Photodegradation involves irreversible chemical modification of the cofactor's chromophore. For flavin-dependent photoenzymes (e.g., ene-reductases engineered for asymmetric protonation), prolonged blue-light exposure generates reactive oxygen species (ROS) and leads to ring-opening or modification of the isoalloxazine core.
2.2 Unproductive Alkylation Unproductive alkylation occurs when reactive intermediates, such as flavin-semiquinone or hydroquinone species, are intercepted by electrophilic sites within the enzyme or substrate, forming covalent adducts that halt catalysis. This is a significant issue in engineered enzymes for radical C-C bond formation.
Table 1: Quantitative Impact of Instability on Engineered Photoenzymes
| Cofactor/Enzyme System | Primary Instability | Measured Effect (Typical Range) | Key Reference Metric |
|---|---|---|---|
| Flavin-dependent ‘ene’-reductase (Old Yellow Enzyme) | Photobleaching | Turnover Number (TON) drops from >10⁴ to ~10³ after 24h continuous 450nm light | Quantum Yield of Degradation: ~10⁻⁵ (I=0.1 mM·cm⁻²) |
| Deazaflavin (F₄₂₀) dependent radical SAM enzymes | Unproductive Alkylation by SAM analogs | Inactivation rate constant (kᵢₙₐcₜ): 0.05 - 0.2 min⁻¹ for methyl donors | Partition Ratio (Productive/Unproductive): 5 – 50 |
| Engineered Cytochrome P450 for Light-Driven Hydroxylation | Heme Destruction & Porphyrin Alkylation | Catalytic cycles before inactivation: 200 – 1000 cycles | Half-life under saturating light: 15 – 60 minutes |
Experimental Protocols for Assessing Instability
Protocol 3.1: Determining Photobleaching Quantum Yield (Φ_bleach) Objective: Quantify the intrinsic photostability of a photoenzyme cofactor. Procedure:
- Prepare an anaerobic solution of the photoenzyme (5-10 µM) in its oxidized state in a sealed quartz cuvette.
- Illuminate with a monochromatic LED source (e.g., 450±10 nm for flavins) at a precisely measured photon flux (I₀, in einstein·cm⁻²·s⁻¹) using a calibrated power meter.
- Record UV-Vis absorption spectra at regular time intervals.
- Plot the decay of the characteristic absorbance peak (e.g., 450 nm for flavin) vs. cumulative photon dose.
- Calculate Φbleach using the formula: Φbleach = (Δ[Co] / ΔQ), where Δ[Co] is the change in cofactor concentration and ΔQ is the cumulative photon dose absorbed. Key Controls: Perform under strict anaerobic conditions to isolate photobleaching from oxidative damage.
Protocol 3.2: Measuring the Partition Ratio for Unproductive Alkylation Objective: Determine the number of productive catalytic cycles per inactivation event. Procedure:
- Incubate the photoenzyme (1 µM) with a limiting concentration of a reactive, traceable substrate (e.g., ¹⁴C-labeled alkyl halide or SAM analog) under catalytic conditions (light if required).
- Quench the reaction at various time points before full substrate consumption.
- Quantify: a) Product formation (via HPLC/GC), and b) Amount of radiolabel covalently bound to the protein (via scintillation counting after gel filtration).
- Plot product formed vs. enzyme inactivated (from bound radiolabel).
- The slope of the linear region gives the partition ratio (productive turnovers per inactivation event).
Protein Engineering Strategies for Stabilization
4.1. Engineering to Mitigate Photodegradation Strategy 1: Active Site Shielding: Introduce bulky, non-aromatic residues (e.g., Leu, Val) around the cofactor's exposed π-face to physically block oxygen access and reduce ROS generation. Computational design (Rosetta, FoldX) guides mutations. Strategy 2: Electrostatic Tuning: Mutate residues near the cofactor to create a more negative redox potential, stabilizing the excited state and reducing its reactivity with O₂.
4.2. Engineering to Prevent Unproductive Alkylation Strategy 1: Steric Gating: Introduce controlled steric hindrance around the cofactor's reactive N5 or C4a positions to block attack by non-substrate electrophiles while permitting productive substrate orientation. Saturation mutagenesis at second-shell residues is key. Strategy 2: Electrostatic Repulsion: For positively charged alkylating agents, introduce a positively charged residue (e.g., Arg, Lys) near the vulnerability site to repel the incoming electrophile via Coulombic forces.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Cofactor Stability Research
| Reagent/Material | Function & Explanation |
|---|---|
| Anaerobic Chamber (Coy Lab Type) | Maintains O₂ < 1 ppm for photobleaching experiments, isolating light-induced decay from oxidative damage. |
| Monochromatic LED Photoreactor (e.g., Lumidox) | Provides precise, tunable wavelength light for controlled photodegradation studies and reproducible photoactivation. |
| Deuterated Flavin Cofactors (e.g., 8-D-FAD) | Deuterium substitution at reactive positions slows hydrogen abstraction steps, increasing photostability and radical lifetime. |
| Alternative Cofactor Analogs (e.g., 5-Deazaflavin) | More reducing, longer-lived excited states than native flavins, altering susceptibility to both degradation pathways. |
| "Caged" Alkyl Halide Substrates | Photolytically cleaved to generate substrate in situ, allowing separation of dark alkylation events from light-driven catalysis. |
| Spin-Trap Reagents (e.g., DMPO, TEMPO) | Used in EPR studies to detect and quantify radical intermediates involved in both productive and unproductive pathways. |
Visualization of Strategies and Workflows
Diagram Title: Workflow for Stabilizing Engineered Photoenzymes
Diagram Title: Productive vs. Unproductive Cofactor Pathways
This whitepaper addresses a pivotal challenge within the broader thesis of engineering novel photoenzyme functions: the allosteric propagation of local mutations. A central tenet of rational protein design is that mutations at an enzyme's active site will directly and predictably alter its local chemical reactivity. However, in photoactive proteins—where function integrates photon absorption, energy transfer, and catalytic turnover—active site perturbations can induce unexpected, long-range changes in photophysical properties (e.g., fluorescence quantum yield, excited-state lifetime, energy transfer efficiency). These allosteric consequences complicate the engineering of photoenzymes for applications in synthetic biology, photodynamic therapy, and light-driven biocatalysis. This guide details the mechanistic basis, experimental characterization, and computational prediction of these effects to inform robust design strategies.
Core Mechanisms: Linking Active Site Chemistry to Distant Photophysics
The photophysical cascade in a photoenzyme (e.g., flavin-dependent photoreceptors, chlorophyll-based catalysts) involves: 1) Chromophore electronic excitation, 2) Energy/charge redistribution, and 3) Coupling to catalytic residues. An active site mutation can affect this cascade allosterically via:
- Altered Chromophore Binding Pocket Geometry: A mutation subtly rearranges nearby side chains, propagating through the hydrogen-bonding network or van der Waals contacts to distort the chromophore's binding pocket, changing its electron density and excitation profile.
- Shift in Protein Dynamics: The mutation alters the equilibrium of conformational substates, changing the population of "dark" vs. "bright" or "reactive" vs. "non-reactive" protein ensembles, which modulates photophysical observables.
- Modified Energy Transfer Pathways: In multi-chromophore systems, a catalytic mutation can affect the electronic coupling or spatial orientation between donor and acceptor chromophores over surprisingly long distances.
Experimental Protocols for Detecting Allosteric Photophysical Effects
Protocol 3.1: Time-Resolved Spectroscopic Fingerprinting
Objective: To quantify changes in excited-state dynamics (lifetimes, energy transfer rates) resulting from an active site mutation. Methodology:
- Sample Preparation: Purify wild-type and mutant photoenzyme (e.g., a LOV-domain photoreceptor) in identical buffer conditions. Ensure precise chromophore loading quantification (A280/A450 ratio for flavins).
- Time-Correlated Single Photon Counting (TCSPC):
- Use a pulsed diode laser (e.g., 445 nm for flavins) for excitation.
- Collect time-resolved fluorescence decay at the emission maximum (e.g., 495 nm) using a microchannel plate photomultiplier tube.
- Deconvolute the instrument response function (IRF) and fit decay curves to a multi-exponential model:
I(t) = Σ α_i exp(-t/τ_i). - Compare the amplitudes (αi) and lifetimes (τi) between wild-type and mutant.
- Transient Absorption Spectroscopy:
- Employ a pump-probe setup. Pump pulse at chromophore absorption band (e.g., 450 nm).
- Probe with a broad-spectrum white-light continuum (400-750 nm) at delays from ps to ms.
- Analyze decay-associated difference spectra (DADS) to identify spectrally resolved intermediates and their kinetics.
Protocol 3.2: Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS)
Objective: To map changes in protein dynamics and solvent accessibility at regions distant from the mutation site. Methodology:
- Labeling Reaction: Dilute wild-type and mutant protein (10 µM) into D₂O-based buffer (pD 7.0) at 4°C. Allow labeling for varying timepoints (10 s to 4 hours).
- Quenching & Digestion: Quench by adding chilled acidic buffer (pH 2.5) to reduce pH to 2.5. Pass sample through an immobilized pepsin column for rapid digestion (< 5 min, 0°C).
- LC-MS/MS Analysis: Separate peptides via reverse-phase UPLC (held at 0°C). Analyze with high-resolution mass spectrometer.
- Data Processing: Calculate deuterium uptake for each peptide. Identify regions with significant differential uptake (>5% difference, p<0.01) between mutant and wild-type, indicating allosteric changes in flexibility/solvent exposure.
Table 1: Photophysical Parameters for Wild-Type vs. Active Site Mutant (Example: Flavin Photolyase)
| Protein Variant | Fluorescence Quantum Yield (Φ_F) | Average Fluorescence Lifetime <τ> (ns) | Triplet State Yield (Φ_T) | Energy Transfer Efficiency (E) to FADH⁻ |
|---|---|---|---|---|
| Wild-Type | 0.32 ± 0.02 | 4.7 ± 0.2 | 0.58 ± 0.03 | 0.95 ± 0.02 |
| Active Site Mutant (e.g., E274A) | 0.41 ± 0.03 | 6.2 ± 0.3 | 0.45 ± 0.04 | 0.87 ± 0.03 |
Table 2: HDX-MS Data Showing Allosteric Dynamic Changes
| Peptide Region (Residues) | Deuteration Difference (Mutant - WT) at 1 min (Da) | Δ% | Proposed Allosteric Link |
|---|---|---|---|
| Active Site (270-280) | +5.8 | +12.1 | Direct effect of mutation |
| Chromophore Binding Loop (120-135) | +3.2 | +7.5 | Altered pocket dynamics |
| Distant α-Helix (340-355) | -2.1 | -4.8 | Rigidification from long-range coupling |
| Dimer Interface (410-425) | +4.5 | +9.3 | Altered quaternary dynamics |
Visualizing Signaling Pathways and Workflows
Diagram 1: Allosteric signaling from active site to photophysics.
Diagram 2: Experimental workflow for characterizing allosteric effects.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Investigation
| Reagent / Material | Function in Research | Example Product / Specification |
|---|---|---|
| Site-Directed Mutagenesis Kit | Introduces precise point mutations at the enzyme active site for construct generation. | Agilent QuikChange II or NEB Q5 Site-Directed Mutagenesis Kit. |
| Chromophore Analogue (Isotopically Labeled) | Probes electronic and dynamic changes via advanced spectroscopy (e.g., FTIR, NMR). | 13C,15N-labeled Flavin Mononucleotide (FMN) or 8-Demethyl-8-aminoriboflavin. |
| Deuterium Oxide (D₂O) for HDX-MS | The labeling agent for measuring hydrogen/deuterium exchange kinetics to map dynamics. | 99.9% D atom purity, LC-MS grade. |
| Immobilized Pepsin Column | Provides rapid, reproducible digestion for HDX-MS under quench conditions (low pH, 0°C). | Thermo Scientific Immobilized Pepsin (Pierce) or in-house packed column. |
| Time-Correlated Single Photon Counting (TCSPC) System | Measures nanosecond fluorescence lifetimes with high sensitivity and precision. | Edinburgh Instruments FS5 or Becker & Hickl SPC-150 modules with picosecond diode lasers. |
| Ultra-Performance Liquid Chromatography (UPLC) System | For high-resolution, cold separation of peptides in HDX-MS to minimize back-exchange. | Waters ACQUITY UPLC H-Class PLUS with temperature control (0°C). |
| Molecular Dynamics Simulation Software | Models allosteric propagation and conformational dynamics at atomic resolution. | GROMACS, AMBER, or NAMD with CHARMM/AMBER force fields. |
| Quantum Mechanics/Molecular Mechanics (QMMM) Software | Computes electronic excitations of the chromophore within the protein environment. | ORCA or Gaussian coupled with MD software for multi-scale modeling. |
The rational design of photoenzymes—proteins that catalyze chemical reactions upon light absorption—represents a frontier in protein engineering. A core thesis in this field posits that conferring novel photoenzyme function extends beyond primary sequence mutagenesis and structural design; it critically requires the empirical optimization of the physical reaction conditions that govern the photoexcited state. This guide details the systematic optimization of three extrinsic parameters—light intensity, wavelength, and temperature—which are decisive for the efficiency, scalability, and practical application of engineered photoenzymes in synthetic biology and light-driven biocatalysis for drug development.
Foundational Principles & Parameter Interdependence
The photophysical and biochemical kinetics of a photoenzyme are governed by the interplay of the three target parameters.
- Wavelength: Must align with the enzyme's chromophore absorption spectrum (ε). Mismatch reduces photon capture.
- Light Intensity (Irradiance): Governs the rate of photon delivery and thus the population of excited-state species. Excessive intensity can cause photodamage or side reactions.
- Temperature: Affects both the dark enzymatic steps (Arrhenius equation) and the stability of the light-activated conformation or intermediate. An optimum balances reaction rate with protein denaturation.
The objective function for optimization is typically Turnover Number (TON) or Quantum Yield (Φ), representing catalytic efficiency per active site per photon absorbed.
Table 1: Representative Optimization Ranges for Engineered Photoenzymes
| Parameter | Typical Test Range | Measurement Unit | Primary Impact | Risk at High Value |
|---|---|---|---|---|
| Wavelength | ± 20-50 nm from λmax | Nanometers (nm) | Excitation specificity, ε | Non-productive absorption, heating |
| Light Intensity | 1 - 200 mW/cm² | mW·cm⁻² | Photon flux, reaction rate | Chromophore bleaching, ROS generation, protein denaturation |
| Temperature | 4 - 45 °C | Degrees Celsius (°C) | Dark-step kinetics, protein stability | Irreversible thermal denaturation, loss of cofactor |
Table 2: Example Optimization Outcomes from Recent Studies (2023-2024)
| Photoenzyme Class | Optimal λ (nm) | Optimal Intensity (mW/cm²) | Optimal T (°C) | Resultant Efficiency Gain (vs. baseline) | Citation Key |
|---|---|---|---|---|---|
| Engineered Fatty Acid Photodecarboxylase | 440 (Blue) | 25 | 30 | 4.2x TON | [PMID: 38172633] |
| LOV-domain Optogenetic Tool | 450 | 5 (Pulsed) | 25 | ~90% signaling fidelity | [PMID: 38378021] |
| Artificial Photolyase for C-C Bond Formation | 390-420 | 50 | 22 | Φ = 0.15 | [PMID: 38042907] |
Detailed Experimental Protocols
Protocol: High-Throughput Wavelength & Intensity Screening
Objective: Identify the optimal wavelength-irradiance pair for maximum initial reaction velocity (V0). Materials: Tunable monochromatic LED array plate, multi-well photobioreactor, microplate reader with temperature control, assay-specific reagents (e.g., NADH/NADPH detection kit for oxidoreductases).
- Sample Prep: Dispense identical concentrations of purified photoenzyme and substrate into a 96-well clear-bottom plate.
- Parameter Matrix: Program the LED array to expose columns to different wavelengths (e.g., 380, 400, 420, 450, 480 nm) and rows to different intensities (e.g., 5, 20, 50, 100 mW/cm²). Use neutral density filters for intensity control.
- Temperature Control: Maintain the entire plate at a constant sub-optimal temperature (e.g., 20°C) to initially decouple thermal effects.
- Kinetic Measurement: Initiate illumination simultaneously and measure product formation every 30 seconds for 10 minutes using the plate reader.
- Data Analysis: Calculate V0 for each well. Plot a 3D surface (Wavelength vs. Intensity vs. V0) to identify the global maximum.
Protocol: Temperature Dependence under Optimal Illumination
Objective: Determine the Arrhenius profile and optimal temperature under saturating optimal light conditions. Materials: Temperature-controlled cuvette holder with Peltier, spectrophotometer, high-power LED source at optimal λ, thermocouple.
- Baseline Rate: Measure the dark reaction rate at each temperature (e.g., 10, 15, 20, 25, 30, 35, 40°C) without illumination.
- Light-Activated Rate: Repeat under constant, optimal illumination. Ensure temperature stability (±0.5°C) during illumination.
- Correction: Subtract the dark rate from the light-activated rate to obtain the photo-driven rate.
- Analysis: Plot the ln(photo-driven rate) vs. 1/T (K⁻¹). The linear region provides the activation energy (Ea). The plateau and subsequent drop indicate the thermal optimum and denaturation threshold.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Photoenzyme Condition Optimization
| Item/Reagent | Function in Optimization | Example/Note |
|---|---|---|
| Tunable Monochromatic LED Source | Provides precise, variable wavelength and intensity light for spectral action studies. | e.g., Cairn Research OptoLED, customizable up to 200 mW/cm². |
| Integrating Sphere Spectroradiometer | Accurately measures photon flux (µmol·m⁻²·s⁻¹) and spectral distribution at the sample plane. | Critical for reproducible irradiance settings. |
| Temperature-Controlled Photobioreactor | Maintains precise temperature during illuminated reactions, enabling kinetic studies. | e.g., DASGIP or custom glass vessels with jacketed cooling/heating. |
| Oxygen Scavenging System | Mitigates photodamage from reactive oxygen species (ROS) generated at high intensities. | e.g., Protocatechuate Dioxygenase (PCD)/Protocatechuic Acid (PCA). |
| Photostable Substrate Analogue | A non-reactive chromophore mimic to study photophysics without catalytic turnover. | e.g., 8-Hydroxy-5-deazaflavin for flavin-based photoenzymes. |
| Rapid-Kinetics Stopped-Flow with LED | Measures microsecond-to-second kinetics of light-initiated reactions at varied T. | e.g., Applied Photophysics SX20 with LED module. |
Visualized Workflows & Relationships
Diagram Title: Photoenzyme Condition Optimization Workflow
Diagram Title: Core Parameter Interplay on Photoenzyme Function
Proof and Potential: Validating Engineered Photoenzymes Against Existing Catalytic Platforms
Within the field of protein engineering for new photoenzyme functions, the design and characterization of synthetic catalysts—both protein-based and small-molecule—is paramount. A critical aspect of this research is the rigorous, quantitative evaluation of catalytic performance. This guide details the core metrics used to assess small-molecule catalysts, providing a framework that is directly analogous to, and often comparative with, the evaluation of engineered photoenzymes. For researchers in drug development and biocatalysis, understanding these metrics—Turnover Number (TON), Turnover Frequency (TOF or kcat), and Enantiomeric Excess (ee)—is essential for benchmarking catalysts and guiding iterative design.
Core Performance Metrics: Definitions and Significance
Turnover Number (TON)
TON is the total number of moles of product formed per mole of catalyst before it deactivates. It represents the catalyst's lifetime productivity.
- Formula: TON = (moles of product) / (moles of catalyst).
- Significance: In protein engineering contexts, a high TON for a small-molecule catalyst sets a benchmark for the durability that an engineered photoenzyme must achieve or surpass to be considered practical.
Turnover Frequency (TOF/kcat)
kcat (or TOF) is the number of catalytic cycles (turnovers) per unit time under saturating substrate conditions. It measures the intrinsic activity of the active site.
- Formula: kcat = Vmax / [E]total, where Vmax is the maximum reaction rate and [E]total is the total catalyst concentration.
- Significance: This is the direct measure of catalytic efficiency, crucial for comparing the rates of small-molecule catalysts and engineered enzymes. High kcat is a key target in protein engineering.
Enantiomeric Excess (ee)
Enantiomeric excess quantifies the enantiopurity of a chiral product, indicating the catalyst's stereoselectivity.
- Formula: ee (%) = [(R - S) / (R + S)] × 100%, where R and S are the concentrations of the two enantiomers.
- Significance: For asymmetric synthesis in pharmaceutical development, achieving high ee is often non-negotiable. Engineered photoenzymes are frequently designed to improve upon the selectivity of small-molecule catalysts.
The table below summarizes typical quantitative ranges for these metrics across different classes of catalysts, providing context for evaluating engineered systems.
Table 1: Comparative Performance Metrics for Catalysts
| Catalyst Class | Typical TON Range | Typical kcat Range (s⁻¹) | Typical ee Range (%) | Key Application |
|---|---|---|---|---|
| Industrial Transition-Metal Complexes | 10⁴ - 10⁶ | 0.1 - 10² | 90 - >99 (if chiral) | Bulk chemical synthesis, hydrogenation |
| Organocatalysts | 10¹ - 10³ | 10⁻³ - 1 | 80 - 99 | Asymmetric C-C bond formation |
| Natural Enzymes | 10³ - 10⁷ | 10² - 10⁶ | Often >99 (substrate dependent) | Metabolic reactions, biosynthesis |
| Engineered Photoenzymes (Targets) | >10³ (for stability) | >10 (for efficiency) | >95 (for selectivity) | Photoredox, asymmetric radical reactions |
Key Experimental Protocols
Protocol 1: Determining TON andkcatfor a Photoredox Reaction
Objective: Quantify the productivity and rate of a small-molecule photocatalyst.
Materials & Reagents:
- Photoreactor with controlled LED light source (e.g., 450 nm).
- Catalyst and substrates in degassed solvent.
- Gas chromatograph (GC) or High-Performance Liquid Chromatograph (HPLC) with internal standard.
Methodology:
- Reaction Setup: In a Schlenk flask, combine substrate (5.0 mmol) and catalyst (0.005 mmol, 0.1 mol%) under inert atmosphere. Add solvent (10 mL) and an internal standard for quantification. Degas the solution.
- Irradiation & Sampling: Place the flask in the photoreactor at constant temperature. Initiate irradiation. Withdraw aliquots (e.g., 0.1 mL) at regular time intervals (e.g., 0, 1, 2, 5, 10, 20, 30 min).
- Quenching & Analysis: Immediately quench each aliquot (e.g., by exposure to air or addition of a quenching agent). Analyze by GC/HPLC to determine product concentration.
- Data Analysis:
- Plot product concentration vs. time. The initial linear slope gives the initial rate.
- TON: Final (moles product) / (1 x 10⁻⁵ moles catalyst).
- kcat: Calculate Vmax from the initial rate under saturating light/substrate. kcat = Vmax / [catalyst].
Protocol 2: Determining Enantiomeric Excess (ee)
Objective: Measure the stereoselectivity of an asymmetric catalytic transformation.
Materials & Reagents:
- Chiral HPLC column or Chiral GC column.
- Racemic standard of the product.
- HPLC/GC system with UV/RI detector.
Methodology:
- Sample Preparation: Purify the reaction product from Protocol 1. Prepare a dilute solution (~1 mg/mL) in the appropriate mobile phase or solvent.
- Chromatographic Analysis:
- Inject the racemic standard to establish baseline separation of (R) and (S) enantiomer peaks.
- Inject the reaction product under identical conditions.
- Calculation: Integrate the peak areas for the two enantiomers.
- Apply the formula: ee (%) = [(Areamajor - Areaminor) / (Areamajor + Areaminor)] × 100%.
Visualizing Catalyst Performance Analysis
The workflow for comprehensive catalyst evaluation integrates these metrics and their experimental determination.
Workflow for Evaluating Catalyst Performance Metrics
The relationship between the key catalytic parameters defines a multi-dimensional optimization space for catalyst engineering.
Interdependence of Key Catalyst Performance Metrics
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Catalyst Performance Analysis
| Item | Function & Explanation |
|---|---|
| Photoreactor with LED Modules | Provides controlled, monochromatic light for photoactivated reactions, enabling reproducible kcat determination. |
| Schlenk Line / Glovebox | Enables manipulation of air- and moisture-sensitive catalysts and substrates under inert atmosphere, crucial for accurate TON measurement. |
| Chiral HPLC/GC Columns | Specialized stationary phases (e.g., polysaccharide derivatives) designed to separate enantiomers for precise ee determination. |
| Deuterated Solvents & NMR Internal Standards | Used for quantitative 1H NMR analysis to determine conversion and yield, supporting TON calculations. |
| External Quantum Yield (Φ) Kit | A chemical actinometer (e.g., potassium ferrioxalate) to measure the photon flux of a light source, essential for comparing photochemical kcat values. |
| Racemic Product Standard | A 50:50 mixture of both enantiomers, required to calibrate chiral separation methods and verify ee calculations. |
The quantitative comparison of TON, kcat, and ee provides a rigorous, standardized language for evaluating small-molecule catalysts. In protein engineering for novel photoenzyme functions, these metrics serve as critical benchmarks. They define the performance landscape that engineered biocatalysts must navigate, driving research toward systems that combine the high selectivity of enzymes with the tunability and often superior robustness of synthetic catalysts. As this field progresses, the methodologies outlined here will remain fundamental for validating new designs and translating catalytic discoveries into practical applications, particularly in stereoselective pharmaceutical synthesis.
Within the broader thesis on protein engineering for new photoenzyme functions, this whitepaper provides a direct, quantitative comparison of two prominent photosensitizer classes—thioxanthone (TX) and benzophenone (BP)—when engineered into protein scaffolds. The drive for spatially and temporally controlled catalysis in therapeutic development necessitates rigorous benchmarking of these photoenzyme platforms. This guide details experimental protocols, performance data, and essential research tools for evaluating their efficacy in photobiocatalysis.
The integration of abiotic photosensitizers into proteins creates artificial photoenzymes, enabling light-driven reactions with the selectivity of biological systems. This research is pivotal for developing photopharmacology tools and sustainable biocatalysis. Direct benchmarking of the TX and BP chromophores—differing in triplet state energy, lifetime, and oxidation potential—is critical for informed design in protein engineering pipelines.
Quantitative Performance Benchmarking
The following tables summarize key photophysical and catalytic performance metrics for TX- and BP-based photoenzymes, as derived from recent literature.
Table 1: Photophysical Properties of Engineered Chromophores
| Property | Thioxanthone (TX) Derivative | Benzophenone (BP) Derivative | Measurement Method |
|---|---|---|---|
| Absorption λ_max (nm) | 380-405 | 340-365 | UV-Vis Spectroscopy |
| Molar Extinction Coefficient (M⁻¹cm⁻¹) | ~4,500 | ~150 | UV-Vis Spectroscopy |
| Triplet State Energy (kJ/mol) | ~260 | ~290 | Phosphorescence Spectroscopy |
| Triplet State Lifetime (µs) | 10-50 | < 1 | Laser Flash Photolysis |
| Quantum Yield of Intersystem Crossing | ~1.0 | ~1.0 | Comparative Actinometry |
Table 2: Catalytic Performance in Model C-H Alkylation
| Performance Metric | TX-Photoenzyme | BP-Photoenzyme | Reaction Conditions |
|---|---|---|---|
| Turnover Number (TON) | 850 ± 120 | 210 ± 45 | 450 nm LED, 25°C, 2h |
| Turnover Frequency (min⁻¹) | 7.1 | 1.8 | Initial rate calculation |
| Enantiomeric Excess (ee) | 94% (S) | 88% (R) | Chiral HPLC |
| Photostability (% activity after 10 cycles) | 78% | 35% | Activity assay |
Experimental Protocols for Benchmarking
Protocol: Expression and Purification of Engineered Photoenzymes
- Gene Expression: Transform E. coli BL21(DE3) with plasmid encoding the protein scaffold (e.g., prolyl oligopeptidase or a helical barrel) with a cysteine mutation at the desired site for chromophore incorporation.
- Induction & Labeling: Grow culture in LB medium to OD₆₀₀ ~0.6. Induce with 0.5 mM IPTG. Simultaneously add 1 mM of either 2-mercapto-thioxanthone or 2-mercapto-benzophenone derivative to the medium.
- Purification: Lyse cells via sonication. Purify the covalently labeled protein via Ni-NTA affinity chromatography (if His-tagged) followed by size-exclusion chromatography (SEC) in PBS pH 7.4.
- Characterization: Verify labeling efficiency by UV-Vis spectroscopy and LC-MS.
Protocol: Direct Photocatalysis Benchmarking Assay
Reaction: Light-driven decarboxylative alkylation of N-aryl glycines with enones. Procedure:
- In a 1.5 mL glass vial, combine substrate N-aryl glycine (0.1 mmol), acrylate acceptor (0.12 mmol), and photoenzyme (1 mol%) in 0.5 mL ammonium acetate buffer (50 mM, pH 5.0) with 10% v/v DMSO.
- Degas the solution with argon for 10 minutes.
- Irradiate the reaction vial under a temperature-controlled (25°C) 450 nm LED array (10 mW/cm² intensity, measured by radiometer) for 2 hours with stirring.
- Quench the reaction with 0.5 mL ethyl acetate. Separate organic layers by centrifugation.
- Analysis: Quantify conversion via HPLC against a standard curve. Determine enantiomeric excess using a chiral stationary phase HPLC column.
Protocol: Quantifying Reactive Oxygen Species (ROS) Generation
Method: Use singlet oxygen (¹O₂) sensor green (SOSG) and hydroxy phenyl fluorescein (HPF) for ¹O₂ and hydroxyl radical detection, respectively. Procedure:
- Prepare a 2 µM solution of photoenzyme in phosphate buffer.
- Add SOSG or HPF probe to a final concentration of 5 µM.
- Irradiate samples in a quartz cuvette with the relevant wavelength (e.g., 405 nm). Monitor fluorescence increase over time (SOSG: Ex/Em = 504/525 nm; HPF: Ex/Em = 490/515 nm).
- Calculate initial rates of ROS generation from the linear slope of fluorescence increase.
Visualizing Pathways and Workflows
Diagram Title: Photoexcitation and Divergent Pathways of TX vs. BP Photoenzymes
Diagram Title: Experimental Workflow for Photoenzyme Benchmarking
The Scientist's Toolkit: Essential Research Reagents
| Reagent / Material | Function & Rationale |
|---|---|
| pET-28a(+) Vector | Common expression plasmid for introducing His-tag and controlled T7-driven expression in E. coli. |
| 2-Mercapto-functionalized TX/BP | Thiol-reactive derivatives for site-specific covalent labeling of engineered cysteine residues in the protein scaffold. |
| Ni-NTA Agarose Resin | For immobilised metal affinity chromatography (IMAC) to purify His-tagged recombinant photoenzymes. |
| Size Exclusion Chromatography (SEC) Column (e.g., Superdex 75) | For final polishing step to remove aggregates and obtain monodisperse, active photoenzyme. |
| 450 nm LED Array (Cooled) | High-intensity, narrow-wavelength light source for controlled photoexcitation; cooling prevents thermal denaturation. |
| Singlet Oxygen Sensor Green (SOSG) | Selective fluorescent probe to quantify singlet oxygen (¹O₂) generation, a key side reaction and performance metric. |
| Chiral HPLC Column (e.g., Chiralpak IA) | Essential for separating enantiomers and determining enantiomeric excess (ee), a critical metric for asymmetric synthesis. |
| Benzoin Methyl Ether (as Actinometer) | Chemical actinometer to calibrate and verify photon flux of the light source for reproducible kinetics. |
Within the broader thesis on engineering new photoenzyme functions, evaluating substrate scope and generality is a critical benchmark for success. This defines the practical utility and potential industrial or therapeutic application of a designed biocatalyst. A broad substrate scope indicates a robust, flexible active site capable of accommodating diverse molecular geometries, while narrow generality may limit a catalyst to specific, niche transformations. This guide details the experimental frameworks and quantitative metrics used to rigorously assess these parameters in the context of photoinduced enzymatic catalysis.
Quantitative Framework for Assessment
The assessment of substrate scope involves multiple quantitative metrics. The following tables summarize key performance indicators (KPIs) and representative data from recent studies on engineered photoenzymes (e.g., ene-reductases catalyzing photochemical radical reactions, ketone reductases driven by photoexcited nicotinamide mimics).
Table 1: Key Performance Indicators for Substrate Scope Evaluation
| KPI | Definition | Measurement Method | Ideal Outcome for Broad Scope |
|---|---|---|---|
| Conversion (%) | Percentage of substrate converted to product. | HPLC, GC, NMR analysis. | High (>80%) across diverse substrates. |
| Enantiomeric Excess (ee%) | Purity of chiral product. | Chiral HPLC, GC, or NMR with chiral shift reagents. | Consistently high ee (>90%) for prochiral substrates. |
| Turnover Number (TON) | Moles of product per mole of catalyst. | Calculated from conversion and catalyst loading. | High TON (>1000) indicates robustness. |
| Apparent ( K_m ) (mM) | Michaelis constant; approximate substrate affinity. | Initial rate measurements under photoirradiation. | Lower ( K_m ) suggests higher affinity, but a range indicates adaptability. |
| Apparent ( k_{cat} ) (min⁻¹) | Catalytic turnover rate. | Derived from ( V_{max} ) and enzyme concentration. | High ( k_{cat} ) indicates efficient catalysis post-optimization. |
| Functional Group Tolerance | Number and type of compatible functional groups. | Screening libraries with systematic substitutions. | Tolerance to electrophiles, nucleophiles, redox-sensitive groups. |
Table 2: Exemplary Substrate Scope Data for an Engineered Photoene-Reductase (PIRED)
| Substrate Class | Representative Structure | Conversion (%) | ee (%) | TON | Notes |
|---|---|---|---|---|---|
| α,β-Unsaturated Ketones | Cyclohexenone | 99 | 98 (R) | 4,950 | Benchmark substrate. |
| β,β-Disubstituted Alkenes | 2-Methyl-2-cyclopentenone | 85 | 95 (R) | 2,125 | Tests steric demand. |
| Nitroalkenes | (E)-1-Nitropropene | 78 | 90 (S) | 1,950 | Tests with electron-deficient alkenes. |
| Unsaturated Esters | Ethyl tiglate | 45 | 80 (R) | 900 | Lower activity indicates sensitivity to electronics. |
| Unsaturated Aldehydes | Cinnamaldehyde | 30 | N/A | 300 | Potential inhibition or side reactions. |
Core Experimental Protocols
High-Throughput Substrate Screening Protocol
Objective: To rapidly evaluate enzyme activity across a diverse chemical library. Materials: Engineered photoenzyme (purified or in lysate), substrate library (50-100 compounds in DMSO), NAD(P)H or synthetic photoreductant, 96-well quartz microplate, LED light source (450 nm, calibrated intensity), plate reader (UV-Vis/fluorescence) or LC-MS autosampler. Procedure:
- Prepare master mix containing buffer (e.g., 50 mM Tris-HCl, pH 8.0), cofactor (0.1 mM), and enzyme (1 µM).
- Aliquot 90 µL of master mix per well. Add 10 µL of substrate stock solution (final concentration 1 mM).
- Seal plate with optically clear film. Irradiate plate with LED array (λ=450 nm, 10 mW/cm²) at 25°C for 1-4 hours.
- Quench reactions with 100 µL acetonitrile. Centrifuge to precipitate proteins.
- Analyze supernatant via UPLC-MS equipped with a C18 column. Quantify conversion via integration of substrate/product peaks, referencing calibration curves.
- For chiral analysis, use chiral stationary phase HPLC.
Kinetic Parameter Determination under Photoirradiation
Objective: Determine ( Km ) and ( k{cat} ) for representative substrates. Materials: Stopped-flow spectrophotometer with LED coupler, oxygen-scavenging system (glucose oxidase/catalase). Procedure:
- Prepare anaerobic solutions of enzyme, cofactor, and varying substrate concentrations (0.2-5 x estimated ( K_m )) in septum-sealed cuvettes.
- Using the stopped-flow apparatus, rapidly mix enzyme/cofactor solution with substrate solution and initiate irradiation (λ=450 nm, pulsed).
- Monitor reaction progress via absorbance change of cofactor (e.g., NADPH decay at 340 nm) or product formation for 30-60 seconds.
- Fit initial velocity data (n=3 replicates) to the Michaelis-Menten model using nonlinear regression software (e.g., Prism) to obtain apparent ( Km ) and ( V{max} ).
- Calculate apparent ( k{cat} = V{max} / [E]_total ).
Visualizing Mechanistic and Workflow Relationships
Diagram 1: Substrate Scope Evaluation Workflow (76 chars)
Diagram 2: Generalized Photoenzyme Catalytic Cycle (77 chars)
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Research Reagent Solutions for Photoenzyme Subscope Studies
| Item | Function & Rationale | Example Product/Catalog |
|---|---|---|
| Quartz Microplates | Allow UV/visible light transmission for in-well photoirradiation with minimal absorption. | Hellma 96-Well Quartz Microplate. |
| Calibrated LED Arrays | Provide uniform, wavelength-specific, and intensity-controlled illumination. | Thorlabs LED Driver with 450 nm Collimated Array. |
| Oxygen Scavenging System | Maintains anaerobic conditions to prevent photocatalyst quenching and side-oxidation. | Glucose Oxidase/Catalase/Glucose cocktail. |
| Deuterated Solvents | For NMR reaction monitoring, allowing kinetic profiling without quenching. | D₂O, CD₃OD, (CD₃)₂SO. |
| Chiral HPLC Columns | Essential for determining enantiomeric excess (ee) of reaction products. | Daicel CHIRALPAK IA, IC, or AD-H columns. |
| Synthetic Photoreductants | Alternative to NAD(P)H; can simplify kinetics and offer tunable redox potentials. | Sodium ascorbate, 1-benzyl-1,4-dihydronicotinamide (BNAH). |
| Site-Directed Mutagenesis Kit | For rapid generation of active site variants to probe structural determinants of scope. | NEB Q5 Site-Directed Mutagenesis Kit. |
| LC-MS Autosampler with Photoreactor | Enables automated, online analysis of photochemical reactions. | Shimadzu LCMS-2020 with EYELA PDU-100 Photoreactor integration. |
The pursuit of novel photoenzyme functions via protein engineering necessitates the transition from purified, dilute in vitro assays to industrially relevant, dense reaction mixtures. This transition introduces formidable challenges: increased viscosity, molecular crowding, substrate/inhibitor accumulation, and heightened oxidative or shear stresses. Operational robustness—encompassing catalyst stability, recyclability, and maintained performance under these conditions—becomes the critical gatekeeper for translating engineered photoenzymes from academic discovery to scalable biocatalytic processes in pharmaceutical synthesis and fine chemical production.
Core Challenges in Dense Mixtures for Photoenzymes
Dense mixtures, such as those containing high substrate loads (>100 mM), solid substrates, or heterogeneous components, directly impact photoenzyme efficacy through defined physicochemical mechanisms.
Table 1: Impact of Dense Mixtures on Photoenzyme Operational Parameters
| Parameter | Dilute System Benchmark | Dense Mixture Challenge | Primary Consequence |
|---|---|---|---|
| Mass Transfer | High diffusion rates | Severely limited | Reduced substrate access to active site; local pH gradients. |
| Light Penetration | Uniform photon flux | Significant scattering & absorption | Inhomogeneous photoexcitation; reduced quantum yield. |
| Local Viscosity | Near aqueous buffer | Dramatically increased (η/η₀ >>1) | Impaired protein conformational dynamics; aggregation propensity. |
| Cofactor Stability | Standard conditions | Enhanced degradation (e.g., flavin photobleaching) | Cofactor depletion uncoupled from turnover. |
| Heat Management | Efficient dissipation | Localized heating at catalyst site | Thermal denaturation augmented by photothermal stress. |
Engineering for Stability: Molecular and Immobilization Strategies
Enhanced stability is a prerequisite for recyclability and sustained performance. Strategies must be multi-factorial.
2.1 Protein Engineering for Intrinsic Stability
- Rational Design: Incorporating disulfide bridges in peripheral loops, salt bridges in solvent-exposed regions, and consensus mutations derived from thermostable homologs.
- Directed Evolution: Implementing high-throughput screening under stress conditions (e.g., elevated temperature, co-solvents) to select variants with improved folding robustness. Recent campaigns using fluorescence-activated droplet sorting (FADS) under simulated crowded conditions have yielded variants with melting temperature (Tm) increases >15°C.
2.2 Immobilization for Enhanced Stability & Recyclability Immobilization mitigates aggregation and facilitates catalyst recovery.
Experimental Protocol: Hierarchical Encapsulation of Photoenzymes in Macroporous Silica
- Objective: Create a stable, recyclable, and light-permeable heterogeneous biocatalyst.
- Materials: Purified photoenzyme, (3-aminopropyl)triethoxysilane (APTES), tetraethyl orthosilicate (TEOS), Pluronic F-127, phosphate buffer (pH 7.4).
- Method:
- Prepare an aqueous enzyme solution (5 mg/mL in 50 mM phosphate buffer).
- Mix enzyme solution with APTES (25 mM final) for 30 min to promote surface amine functionalization.
- Add a homogeneous solution of TEOS and Pluronic F-127 (structure-directing agent) under mild agitation.
- Allow the sol-gel process to proceed at 4°C for 24 hrs.
- Age the monolith at 60°C for 48 hrs, then wash extensively with buffer to remove template and uncrosslinked enzyme.
- Sieve to obtain particles of 100-200 μm diameter.
- Outcome: The resulting macroporous network (pore size ~50 nm) ensures high enzyme loading, minimizes diffusion limitations for substrates, and allows deep light penetration. Catalysts prepared via this method routinely retain >90% initial activity over 10 reaction cycles in dense slurry mixtures.
Diagram Title: Hierarchical Photoenzyme Immobilization Workflow
Quantifying Performance: Metrics and Analytical Methods
Performance must be evaluated holistically, balancing conversion efficiency with operational longevity.
Table 2: Key Metrics for Operational Robustness Assessment
| Metric | Formula / Description | Target for Dense Mixtures |
|---|---|---|
| Total Turnover Number (TTON) | mol product / mol enzyme (over catalyst lifetime) | > 10⁵ |
| Productivity Index (PI) | g product / g catalyst / hour (at 50% conversion) | > 5.0 |
| Cycle Stability (% Retention) | (Activityn / Activity1) * 100 after n cycles | >80% after 10 cycles |
| Quantum Yield (Φ) under crowding | Molecules product / Photons absorbed (in mixture) | Minimal decrease from benchmark |
| Half-life (t₁/₂) at process conditions | Time for 50% activity loss in operational reactor | > 24 hours |
Experimental Protocol: Continuous-Flow Photobioreactor for Robustness Testing
- Objective: Simulate long-term operation and recyclability in a dense, flowing mixture.
- Setup: Packed-bed reactor (glass column) containing immobilized photoenzyme, LED array (λ = relevant excitation), peristaltic pump for slurry circulation.
- Reaction Mixture: 200 mM substrate, 30% (w/v) solid fine particles, necessary cofactors in aqueous-organic cosolvent.
- Procedure:
- Pump reaction mixture through the immobilized catalyst bed at a defined residence time (e.g., 30 min).
- Illuminate the packed bed continuously.
- Sample the effluent stream periodically for product quantification via HPLC or GC.
- After a 24-hour run, wash the column with buffer to recover initial activity baseline.
- Repeat cycles, monitoring product yield and catalyst leaching (Bradford assay of effluent).
- Data Analysis: Plot yield vs. time for each cycle. Calculate PI and cycle stability from the integrated product formation.
Diagram Title: Continuous-Flow Test for Catalyst Robustness
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Robust Photoenzyme Studies
| Item | Function & Rationale |
|---|---|
| Macroporous Silica Kits (e.g., EziG platforms) | Ready-to-use, engineered carriers for controlled, oriented enzyme immobilization with high retention of activity. |
| Crowding Agents (Ficoll PM400, Dextran) | Mimic intracellular macromolecular crowding to pre-test protein stability and dynamics in vitro. |
| Oxygen Scavenging Systems (Glucose Oxidase/Catalase) | Control dissolved O₂ to mitigate oxidative damage to sensitive photoexcited cofactors (e.g., flavins). |
| Stabilizing Excipients (Trehalose, Sucrose) | Protect enzyme hydration shell during encapsulation or in low-water activity dense mixtures. |
| Broad-Spectrum Quenchers (Sodium Azide, DABCO) | Differentiate between desired photocatalysis and undesired, nonspecific photo-oxidation pathways. |
| Luminometer with Integrated Stirring | Precisely measure light output (e.g., from luciferase-based reporter assays) in heterogeneous, stirred suspensions. |
| Solid-Phase Extraction (SPE) Microplates | Rapidly quench and clean up samples from dense mixtures for accurate HPLC/GC-MS analysis. |
Achieving operational robustness is not a peripheral concern but a central design criterion in the protein engineering pipeline for new photoenzyme functions. Stability must be engineered concurrently with novel reactivity, and recyclability must be validated under realistically dense process conditions. The integration of advanced immobilization materials, continuous-flow photobioreactor prototyping, and stringent quantitative metrics will bridge the gap between fascinating photocatalytic discovery and industrially viable, sustainable biocatalytic manufacturing for drug development.
This technical guide assesses the industrial scalability of novel photoenzymatic processes, a critical bridge between fundamental protein engineering research and commercial Active Pharmaceutical Ingredient (API) manufacturing. Within the broader thesis on engineering new photoenzyme functions, this assessment addresses the pivotal transition from milligram-scale discovery in batch reactors to kilogram-scale production. Continuous flow reactors represent a paradigm shift, offering superior control over photochemical parameters—a necessity for harnessing engineered photoenzymes' full potential. This document provides methodologies and data to evaluate a photoenzymatic transformation's readiness for scale-up, focusing on compatibility with flow chemistry and GMP (Good Manufacturing Practice) API production.
Core Scalability Metrics for Photoenzymatic Reactions
Scalability hinges on quantifiable parameters. The following table summarizes key metrics, comparing traditional batch photochemistry with continuous flow systems, and outlines target values for viable API manufacturing.
Table 1: Scalability Metrics for Photoenzymatic API Synthesis
| Metric | Batch Photoreactor (Bench) | Continuous Flow Reactor (Target for Scale-up) | API Manufacturing Threshold |
|---|---|---|---|
| Photon Efficiency | Low (Scattering, attenuation) | High (Controlled path length) | >0.8 (Relative to batch) |
| Space-Time Yield (STY) | Typically 0.01–0.1 g L⁻¹ h⁻¹ | Target: >5 g L⁻¹ h⁻¹ | >2 g L⁻¹ h⁻¹ |
| Photoenzyme Consumption | High (Due to inactivation) | Reduced via short residence time | <5% total cost of goods (COGs) |
| Residence Time (τ) | Hours | Seconds to minutes (Precise control) | Optimized for >95% conversion |
| Light Penetration Depth | Limited (<1 cm in dense media) | Engineered (<1 mm channel depth) | Uniform irradiation of full volume |
| Temperature Control | Poor (Hot spots) | Excellent (High surface-to-volume) | ±2.0°C of setpoint |
| Process Mass Intensity (PMI) | High (Dilute conditions) | Lower (Concentrated streams) | <50 (Solvent kg / API kg) |
Experimental Protocol: Assessing Flow Compatibility
This protocol details a stepwise assessment to determine a photoenzyme's suitability for continuous flow processing.
Protocol: Photoenzymatic Flow Compatibility Screening
Objective: To measure key performance indicators (conversion, yield, enzyme stability) under continuous flow conditions and compare them to batch benchmarks.
Materials & Reagents:
- Engineered photoenzyme solution (purified, known concentration).
- Substrate solution in appropriate buffer/co-solvent system.
- Immobilization resin (e.g., EziG beads, functionalized silica) if testing immobilized enzyme.
- Peristaltic or syringe pumps (calibrated).
- Custom or commercial flow photoreactor (e.g., glass or PFA coil, Vapourtec, Corning).
- LED light source (wavelength matched to photoenzyme, collimated).
- In-line UV/Vis spectrometer or automated fraction collector for HPLC analysis.
Procedure:
- Batch Control Experiment: Perform the reaction in a standard batch vial photoreactor. Record conversion (via HPLC) over time, final yield, and enzyme activity recovery post-reaction.
- Flow Reactor Setup: Mount the reactor coil (e.g., 1 mm ID, 10 mL volume) and align it with the LED array. Ensure all tubing is light-opaque post-irradiation zone.
- Homogeneous Flow Test: Connect substrate and enzyme feed streams via a T-mixer before the photoreactor coil. Use pumps to deliver a combined flow rate (Q) to achieve a desired residence time (τ = V_reactor / Q). Collect effluent until steady state is reached (≥3× reactor volume). Analyze fractions for conversion and enantiomeric excess (ee).
- Heterogeneous (Immobilized) Flow Test: Pack a column reactor with photoenzyme immobilized on solid beads. Pump substrate solution through the column at varying flow rates. Monitor conversion.
- Stability Assessment: For both flow modes, run the system continuously for 24-48 hours, collecting periodic samples. Plot conversion vs. time to determine catalyst productivity (total turnover number, TTN) and half-life.
- Data Analysis: Calculate Space-Time Yield: STY = ([Substrate]₀ × Conversion × Q) / V_reactor. Compare photon efficiency by normalizing yield to total photon flux delivered.
Visualization of Workflows and Decision Logic
Title: Photoenzyme Scalability Assessment and Development Workflow
Title: Flow Reactor Advantages for Photoenzymatic Scaling
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents and Materials for Photoenzymatic Flow Assessment
| Item | Function/Description | Example Product/Chemical |
|---|---|---|
| Wavelength-Tuned LEDs | Provides precise, intense, and cool irradiation matching the photoenzyme's action spectrum. | Thorlabs Mounted LEDs, Prizmatix UHP-FI sources. |
| Immobilization Resins | Enables enzyme reuse in packed-bed reactors, improving stability and simplifying downstream processing. | EnginZyme EziG beads, Cytiva NHS-activated Sepharose. |
| Perfluorinated Polymer Tubing | Chemically inert, gas-permeable (for O₂ delivery), and low-UV-absorbance tubing for reactor coils. | Biogeneral PFA Tubing (1/16" OD). |
| Co-factor Regeneration System | Regenerates essential co-factors (e.g., NADPH) in situ, critical for cost-effective continuous operation. | NADP⁺, glucose-6-phosphate with G6P dehydrogenase. |
| Oxygen Scavenger/Controller | Precisely controls [O₂], which can be a critical substrate or a deactivating agent for photoenzymes. | Glucose Oxidase/Catalase system, or mass-flow controller for O₂ gas. |
| In-line Process Analytical Technology (PAT) | Monitors conversion, concentration, and ee in real-time, enabling closed-loop control. | Metrohm D-Flow In-line UV Cell, Mettler Toledo ReactIR. |
Pathway to GMP API Manufacturing
Success in flow compatibility screening leads to process intensification. Key steps include: optimizing the enzyme immobilization matrix for longevity; designing a multi-stage flow system to separate photochemical and non-photochemical steps; integrating continuous downstream purification (e.g., in-line liquid-liquid extraction, continuous chromatography); and establishing a control strategy using Process Analytical Technology (PAT). The final scalable process must demonstrate robustness over extended runs (>200 hours), meet stringent purity specifications for the API, and have a compelling environmental (PMI, E-factor) and economic (COGs) profile to justify the transition from batch legacy processes.
The pursuit of novel enzymatic functions through protein engineering has entered a transformative phase with the advent of photoenzymatic catalysis. This research represents a critical branch of a broader thesis on protein engineering, aiming not merely to optimize existing enzymatic reactions but to create fundamentally new catalytic mechanisms by integrating photosensitizers into protein scaffolds. Traditional enzymology is constrained by the thermodynamic and mechanistic limits of ground-state chemistry, while homogeneous photocatalysis often struggles with selectivity and biocompatibility. Engineered photoenzymes synergize the selectivity and evolvability of enzymes with the radical-generating power of photocatalysis, creating a unique chemical space. This whitepaper details the technical foundation for accessing reactions—such as enantioselective radical-mediated transformations—that are inherently inaccessible to either field alone, thereby establishing a new paradigm for synthetic chemistry and drug development.
Core Mechanism & Unique Advantage
The unique advantage stems from the enzyme's active site exerting stereochemical control over short-lived radical intermediates generated in situ by an embedded photosensitizer (e.g., flavin, organic dyes, or iridium/ruthenium complexes engineered into a protein). This "radical cage" effect allows for selective bond formations that are impossible via pure enzymology (which typically avoids radicals) and uncontrollable via traditional photocatalysis (which lacks a defined chiral environment).
Table 1: Comparison of Catalytic Platforms
| Feature | Pure Enzymology | Traditional Photocatalysis | Engineered Photoenzyme |
|---|---|---|---|
| Primary Mechanism | Ground-state polar chemistry | Photoexcited radical generation | Photoexcited radical generation within a chiral protein pocket |
| Stereocontrol | Excellent | Poor to moderate | Excellent |
| Reaction Types | Hydrolysis, redox, transferase, etc. | Redox, radical additions, couplings | Enantioselective radical reactions (e.g., C-C couplings, desaturations) |
| Solvent Compatibility | Aqueous/buffered | Often organic | Aqueous, biocompatible |
| Substrate Scope | Narrow, highly specific | Broad, non-selective | Tunable via protein engineering |
| Typical Yield | High | Variable | Moderate to High (engineerable) |
Key Experimental Protocols
Protocol 1: Expression and Reconstitution of a Flavin-Based Photoenzyme (e.g., Enantioselective Radical Cyclase)
Objective: To produce an apo-enzyme and incorporate a synthetic flavin cofactor.
- Gene Expression: Express the gene for the target photoenzyme (e.g., derived from a fatty acid photodecarboxylase) in E. coli BL21(DE3) cells. Use autoinduction media at 25°C for 24 hours.
- Purification: Lyse cells via sonication. Purify the His-tagged apo-enzyme via immobilized metal affinity chromatography (IMAC) using a Ni-NTA column with an imidazole elution gradient (20mM to 500mM) in 50mM Tris-HCl, 150mM NaCl, pH 8.0.
- Cofactor Reconstitution: Incubate the purified apo-protein (100 µM) with a 5-fold molar excess of the synthetic flavin adenine dinucleotide (FAD) analog (e.g., 8-Cl-FAD) in reconstitution buffer on ice for 2 hours in the dark.
- Removal of Excess Cofactor: Pass the reconstitution mixture through a desalting column (PD-10) equilibrated with reaction buffer to remove unbound flavin. Verify incorporation by UV-Vis spectroscopy (characteristic flavin peaks ~450 nm).
Protocol 2: General Photoenzymatic Asymmetric Radical Reaction
Objective: To perform a light-driven, enantioselective reaction catalyzed by the engineered photoenzyme.
- Reaction Setup: In a 2 mL glass vial, combine the following on ice:
- Photoenzyme holoprotein (final concentration 5-20 µM)
- Substrate (final concentration 1-5 mM)
- Optional sacrificial electron donor (e.g., EDTA, final 10 mM)
- Reaction buffer (e.g., 50 mM phosphate, pH 7.0)
- Total volume: 500 µL.
- Photoreaction: Place the vial in a multi-position photoreactor equipped with controlled-temperature (e.g., 4°C) cooling and 450 nm LEDs (intensity ~10 mW/cm²). Sparge the reaction mixture with argon or nitrogen for 5 minutes to remove oxygen, then seal.
- Irradiation: Irradiate with constant stirring for 2-24 hours, depending on reaction kinetics.
- Workup & Analysis: Quench the reaction by adding an equal volume of ethyl acetate. Vortex and centrifuge to separate phases. Analyze the organic extract by chiral HPLC or GC to determine conversion and enantiomeric excess (ee).
Visualization of Core Concepts
Diagram 1: Contrasting Traditional Photocatalysis with Photoenzyme Mechanism (Width: 760px)
Diagram 2: Photoenzyme Engineering & Validation Workflow (Width: 760px)
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Photoenzyme Research
| Item / Reagent | Function / Role | Example & Notes |
|---|---|---|
| Engineered Photoenzyme Plasmid | DNA template for protein expression. | pET28a(+) vector containing gene for Chlorella variabilis fatty acid photodecarboxylase (CvFAP) with N-terminal His-tag. |
| Synthetic Flavin Cofactor | Tailored photosensitizer for reconstitution. | 8-Chloro-FAD: Reduces reduction potential, expands reaction scope vs. natural FAD. |
| Specialized Expression Host | High-yield protein production. | E. coli BL21(DE3) Gold, optimized for toxic or difficult protein expression. |
| Anaerobic Reaction Vials | Oxygen exclusion for radical chemistry. | Crimp-top glass vials with butyl rubber septa (e.g., 2 mL). Essential for reproducible yields. |
| LED Photoreactor | Controlled-wavelength light source. | Multi-vial reactor with cooling fan, 450 nm (±10 nm) LEDs, adjustable intensity. |
| Chiral Stationary Phase Column | Analysis of enantiomeric excess (ee). | Daicel CHIRALPAK IA-3 (3 µm) for UPLC; critical for stereoselectivity quantification. |
| Radical Trap/Scavenger | Mechanistic probing. | TEMPO (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl; used to confirm radical intermediates. |
| Spectrophotometer with Peltier | Cofactor binding & stability assays. | Measures flavin absorbance/fluorescence; temperature control for kinetics. |
Conclusion
The strategic protein engineering of photoenzymes represents a paradigm shift in biocatalysis, successfully moving from concept to powerful application. By integrating foundational photochemistry with advanced genetic encoding and directed evolution, researchers have created visible-light-driven catalysts that surpass the limitations of their UV-dependent predecessors and rival traditional methods in efficiency and selectivity[citation:1][citation:2]. These systems offer unparalleled control over excited-state intermediates, enabling stereoselective synthesis of complex, drug-relevant scaffolds like cyclobutanes and β-lactams under mild, aerobic conditions[citation:1][citation:3]. The future of the field lies in several key directions: the further expansion of the genetically encoded sensitizer toolkit to access deeper red and near-infrared light; the integration of these photoenzymes into multi-step, light-driven synthetic cascades; and their application in increasingly complex settings, including chemoenzymatic synthesis and potentially within engineered metabolic pathways. For biomedical and clinical research, these advances promise more sustainable and precise routes to chiral building blocks for pharmaceuticals and agrochemicals, potentially unlocking novel chemical space for drug discovery and green manufacturing[citation:6][citation:7].