Research Articles
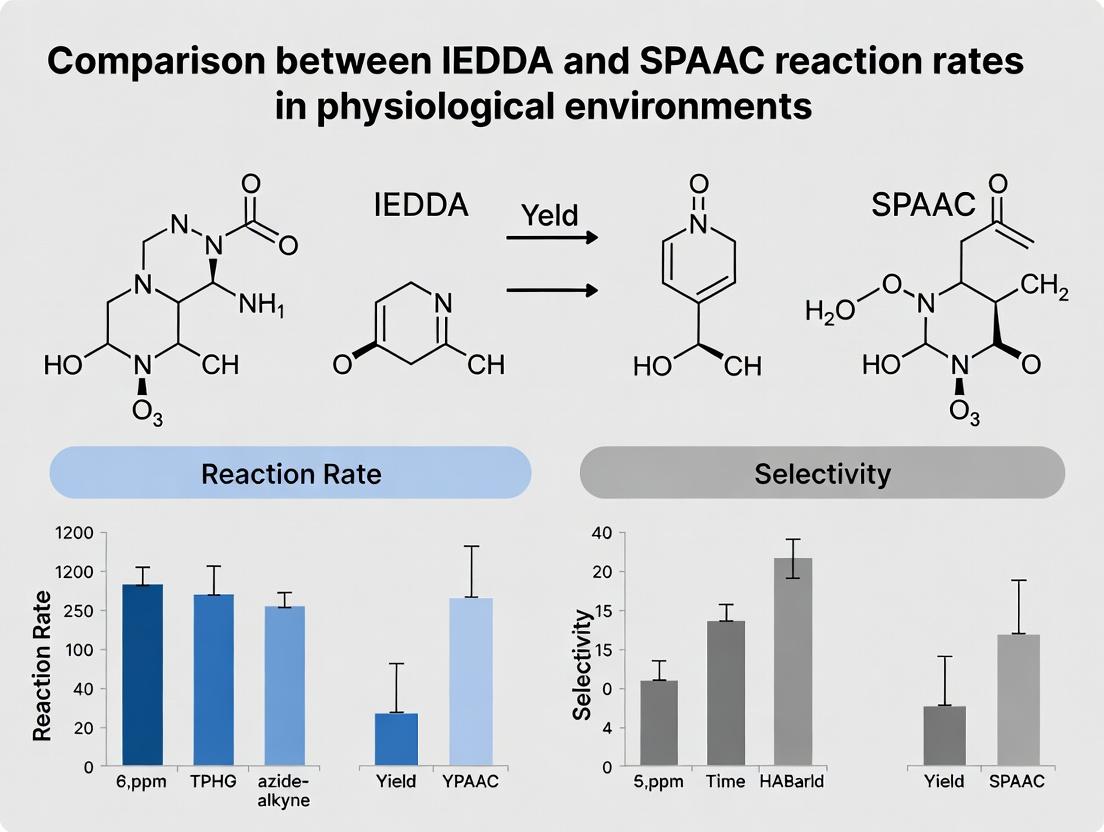
IEDDA vs. SPAAC Click Chemistry: A Comparative Analysis of Reaction Rates in Physiological Environments for Drug Development
This article provides a comprehensive comparative analysis of the reaction kinetics for IEDDA (Inverse Electron-Demand Diels-Alder) and SPAAC (Strain-Promoted Azide-Alkyne Cycloaddition) bioorthogonal click chemistry reactions under physiological conditions.
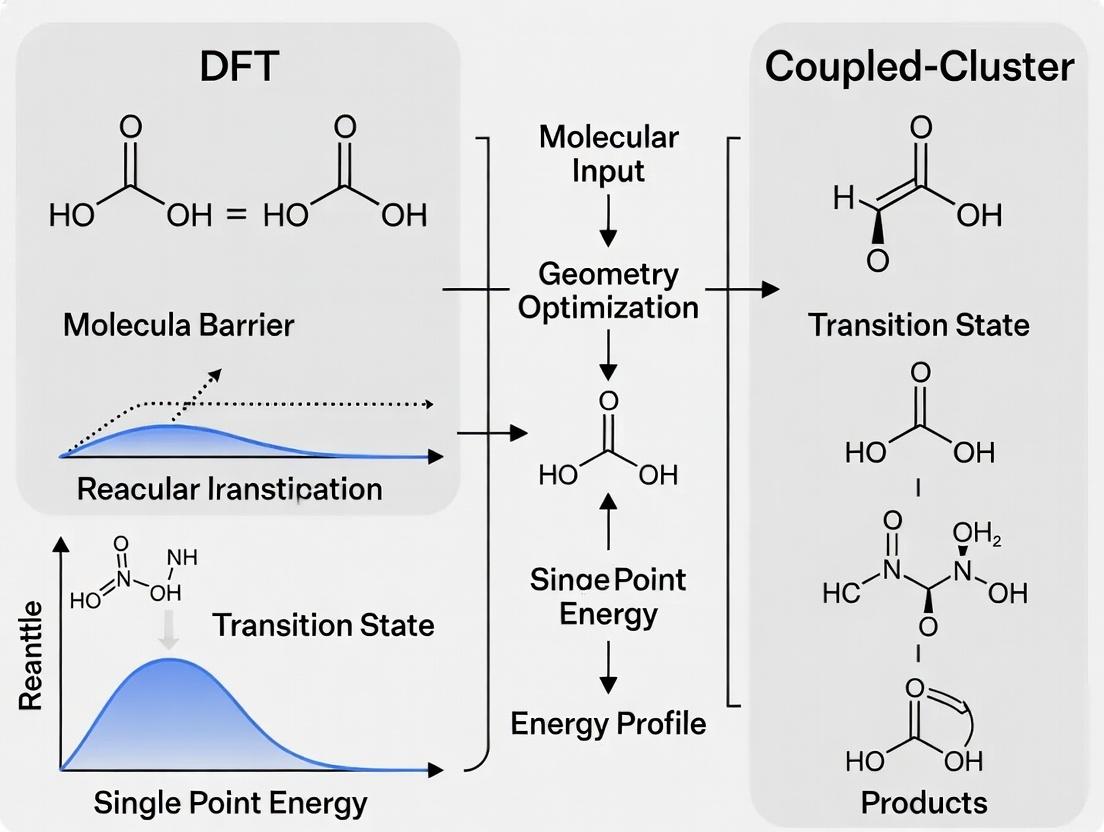
Benchmarking Reaction Barriers: When to Use DFT vs. Coupled-Cluster in Drug Discovery & Catalysis
This article provides a comprehensive guide for computational chemists and drug development researchers on selecting and applying Density Functional Theory (DFT) and coupled-cluster (CC) methods for calculating reaction energy barriers.
Validating DFT Predictions with Spectroscopic Data: A Practical Guide for Computational Chemists and Drug Developers
This article provides a comprehensive guide for researchers, scientists, and drug development professionals on validating Density Functional Theory (DFT) calculations using spectroscopic properties.
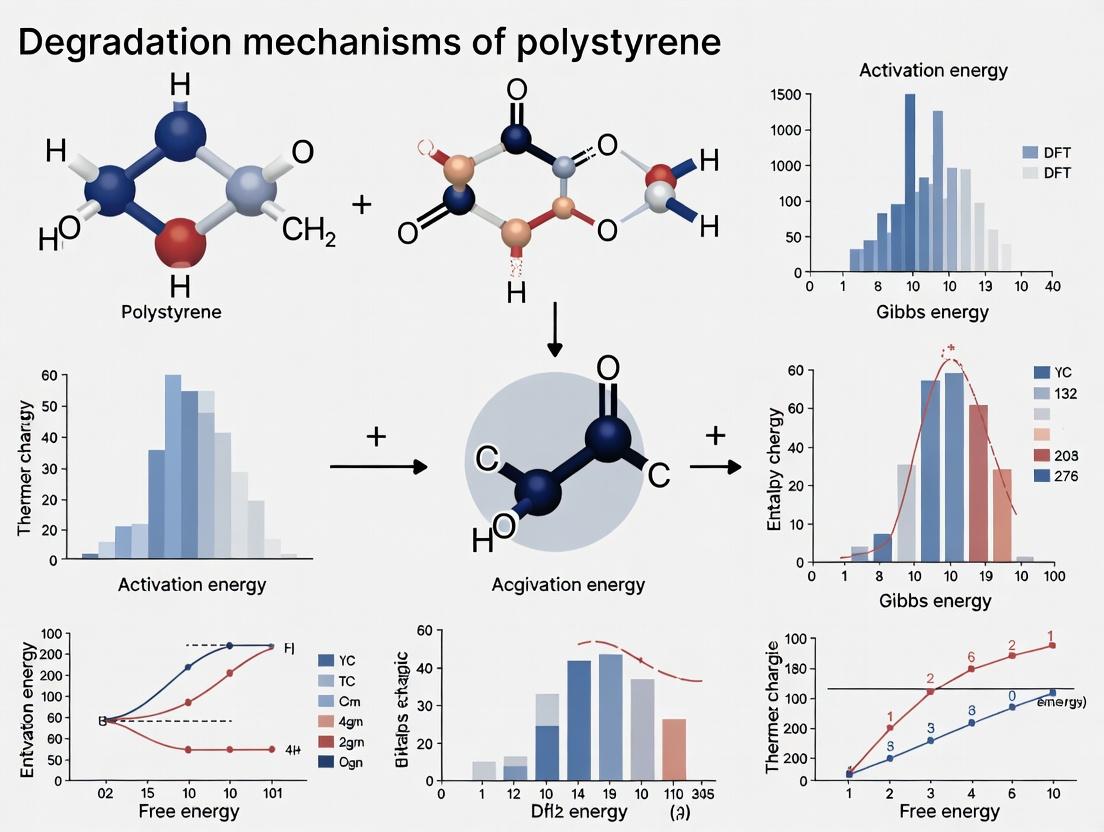
Unraveling Polystyrene Breakdown: A Comprehensive DFT Study on Degradation Mechanisms and Pathways
This article presents a detailed Density Functional Theory (DFT) investigation into the molecular-level degradation mechanisms of polystyrene.
Unlocking 1,3-Dipolar Cycloaddition Mechanisms: A Modern DFT Study Guide for Drug Discovery
This comprehensive article leverages the latest Density Functional Theory (DFT) research to dissect the intricate mechanisms of 1,3-dipolar cycloaddition reactions.
Unlocking Drug Design: A Beginner's Guide to DFT for Reaction Mechanism Analysis
This comprehensive guide introduces Density Functional Theory (DFT) as a pivotal tool for elucidating reaction mechanisms in drug discovery.
Mastering Transition State Optimization in Drug Discovery: A Modern DFT Protocol Guide for Researchers
This article provides a comprehensive guide for computational chemists and drug development researchers on implementing robust Density Functional Theory (DFT) protocols for transition state (TS) optimization.
Decoding Drug Degradation: A DFT Protocol Guide for Catalytic Pathway Analysis in Pharmaceutical Research
This article provides a comprehensive guide to Density Functional Theory (DFT) protocols for elucidating catalytic degradation pathways of pharmaceutical compounds.
Bridging the Accuracy-Cost Gap: Achieving DFT-Level Precision with Semi-Empirical Computational Methods in Drug Discovery
This article explores the emerging paradigm of leveraging modern semi-empirical quantum mechanical (SQM) methods to achieve density functional theory (DFT)-level accuracy at a fraction of the computational cost.
Unlocking Molecular Interactions: A Comprehensive DFT Guide to Adsorption Mechanisms on Surfaces for Biomedical Research
This article provides a detailed, current guide to using Density Functional Theory (DFT) for investigating adsorption mechanisms on material surfaces, tailored for researchers and drug development professionals.